Diagonalizzazione quantistica basata su campioni di un Hamiltoniano chimico
Stima di utilizzo: meno di un minuto su un processore Heron r2 (NOTA: questa è solo una stima. Il tempo effettivo potrebbe variare.)
Obiettivi di apprendimento
Dopo aver completato questo tutorial, gli utenti dovrebbero comprendere:
- Come usare il componente aggiuntivo SQD per Qiskit per approssimare l'energia dello stato fondamentale di un sistema molecolare usando bitstring campionati da un'unità di elaborazione quantistica (QPU).
- Come usare ffsim per costruire un circuito ansatz local unitary cluster Jastrow (LUCJ) per la simulazione di chimica quantistica.
Prerequisiti
Prima di procedere con questo tutorial, raccomandiamo agli utenti di familiarizzarsi con i seguenti argomenti:
- Chimica quantistica e seconda quantizzazione
- Utilizzo della primitiva Sampler per campionare da circuiti quantistici
Background
In questo tutorial, mostriamo come post-elaborare campioni quantistici rumorosi per approssimare lo stato fondamentale della molecola di azoto alla lunghezza di legame di equilibrio, usando il componente aggiuntivo SQD per Qiskit per implementare l'algoritmo di diagonalizzazione quantistica basata su campioni (SQD). Maggiori dettagli sul software si trovano nella documentazione corrispondente, incluso un esempio semplice per iniziare.
Questo tutorial è consigliato agli utenti che hanno familiarità con la chimica quantistica: in particolare, la familiarità con il calcolo delle energie degli stati fondamentali di una molecola. Per una spiegazione dettagliata del flusso di lavoro, consulta il corso sull'algoritmo di diagonalizzazione quantistica.
SQD è una tecnica per trovare autovalori e autovettori di operatori quantistici, come l'Hamiltoniano di un sistema quantistico, usando insieme il calcolo quantistico e il calcolo classico distribuito. Il calcolo classico distribuito viene usato per elaborare campioni ottenuti da un processore quantistico, e per proiettare e diagonalizzare un Hamiltoniano target in un sottospazio da essi generato. Un flusso di lavoro basato su SQD prevede i seguenti passaggi:
- Scegli un ansatz per il circuito e applicalo su un computer quantistico a uno stato di riferimento (in questo caso, lo stato di Hartree-Fock).
- Campiona bitstring dallo stato quantistico risultante.
- Esegui la procedura di recupero della configurazione auto-consistente sui bitstring per ottenere l'approssimazione dello stato fondamentale.
SQD funziona bene quando l'autostato target è sparso: la funzione d'onda è supportata da un insieme di stati di base la cui dimensione non cresce esponenzialmente con la dimensione del problema.
Chimica quantistica
L'Hamiltoniano di un sistema molecolare può essere scritto come
dove e sono numeri complessi chiamati integrali molecolari che possono essere calcolati dalla specifica della molecola usando un programma per computer. In questo tutorial, calcoliamo gli integrali usando il pacchetto software PySCF.
Per i dettagli su come viene derivato l'Hamiltoniano molecolare, consulta un libro di testo di chimica quantistica (ad esempio, Modern Quantum Chemistry di Szabo e Ostlund). Per una spiegazione di alto livello su come i problemi di chimica quantistica vengono mappati sui computer quantistici, guarda la lezione Mapping Problems to Qubits dalla Qiskit Global Summer School 2024.
Ansatz local unitary cluster Jastrow (LUCJ)
SQD richiede un ansatz per il circuito quantistico da cui campionare. In questo tutorial, useremo l'ansatz local unitary cluster Jastrow (LUCJ) per la sua combinazione di motivazione fisica e compatibilità con l'hardware. Useremo ffsim per costruire il circuito ansatz.
L'ansatz LUCJ si adatta alle QPU con connettività dei qubit limitata. Gli orbitali di spin sono mappati sui qubit in modo tale che l'ansatz non richieda routing con gate SWAP. L'hardware IBM® ha una topologia di qubit a reticolo heavy-hex, nel qual caso possiamo adottare un pattern a "zig-zag", illustrato di seguito. In questo pattern, gli orbitali con lo stesso spin sono mappati su qubit con una topologia a linea (cerchi rossi e blu), e una connessione tra orbitali di spin diverso è presente ogni 4° orbitale spaziale, con la connessione facilitata da un qubit ancilla (cerchi viola).
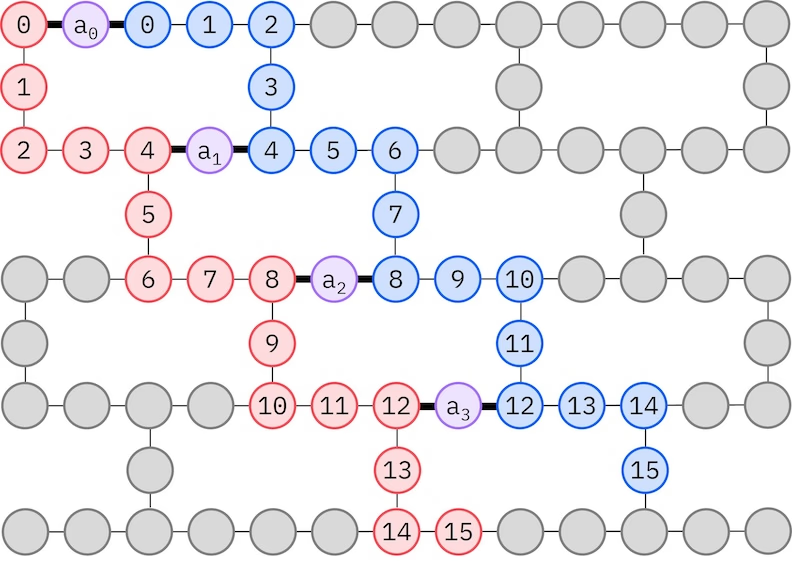
Recupero della configurazione auto-consistente
La procedura di recupero della configurazione auto-consistente è progettata per estrarre il massimo segnale possibile dai campioni quantistici rumorosi. Poiché l'Hamiltoniano molecolare conserva il numero di particelle e lo spin Z, ha senso scegliere un ansatz per il circuito che conservi anch'esso queste simmetrie. Quando applicato allo stato di Hartree-Fock, lo stato risultante ha un numero di particelle fisso e spin Z fisso nel caso senza rumore. Pertanto, le metà spin- e spin- di qualsiasi bitstring campionato da questo stato dovrebbero avere lo stesso peso di Hamming dello stato di Hartree-Fock. A causa della presenza di rumore nei processori quantistici attuali, alcuni bitstring misurati violeranno questa proprietà. Una forma semplice di post-selezione scarterebbe questi bitstring, ma ciò è dispendioso perché quei bitstring potrebbero contenere ancora qualche segnale. La procedura di recupero auto-consistente tenta di recuperare parte di quel segnale nella post-elaborazione. La procedura è iterativa e richiede come input una stima delle occupazioni medie di ciascun orbitale nello stato fondamentale, calcolata inizialmente dai campioni grezzi. La procedura viene eseguita in un ciclo, e ogni iterazione prevede i seguenti passaggi:
- Per ogni bitstring che viola le simmetrie specificate, capovolgi i suoi bit con una procedura probabilistica progettata per avvicinare il bitstring alla stima corrente delle occupazioni medie degli orbitali, per ottenere un nuovo bitstring.
- Raccogli tutti i bitstring vecchi e nuovi che soddisfano le simmetrie, e campiona sottoinsiemi di dimensione fissa, scelta in anticipo.
- Per ogni sottoinsieme di bitstring, proietta l'Hamiltoniano nel sottospazio generato dai vettori di base corrispondenti (vedi la sezione precedente per una descrizione di questi vettori di base), e calcola una stima dello stato fondamentale dell'Hamiltoniano proiettato su un computer classico.
- Aggiorna la stima delle occupazioni medie degli orbitali con la stima dello stato fondamentale con l'energia più bassa.
Diagramma del flusso di lavoro SQD
Il flusso di lavoro SQD è illustrato nel seguente diagramma:
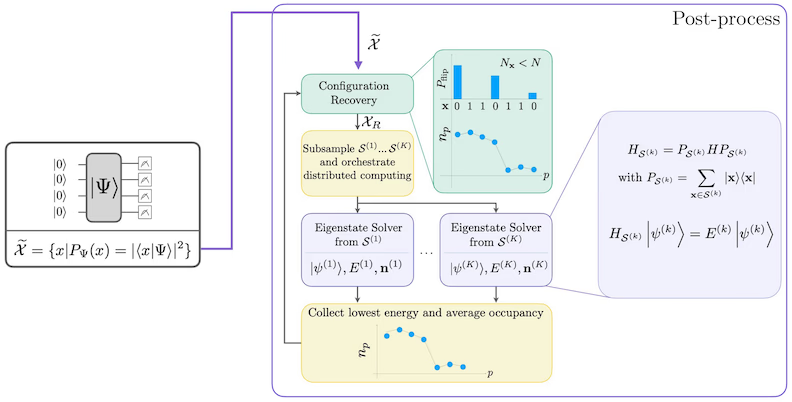
Requisiti
Prima di iniziare questo tutorial, assicurati di avere installato quanto segue:
- Qiskit SDK v1.0 o successivo, con supporto per la visualizzazione
- Qiskit Runtime v0.22 o successivo (
pip install qiskit-ibm-runtime) - Componente aggiuntivo SQD per Qiskit v0.11 o successivo (
pip install qiskit-addon-sqd) - ffsim v0.0.75 o successivo (
pip install ffsim)
Configurazione
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import math
import ffsim
import matplotlib.pyplot as plt
import numpy as np
import pyscf
import pyscf.cc
import pyscf.mcscf
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.primitives import StatevectorSampler
from qiskit.providers.fake_provider import GenericBackendV2
from qiskit_ibm_runtime import QiskitRuntimeService
from qiskit_ibm_runtime import SamplerV2 as Sampler
Esempio su simulatore su piccola scala
In questo tutorial, troveremo un'approssimazione dello stato fondamentale di una molecola di azoto vicino alla sua distanza di legame di equilibrio. Prima usiamo un piccolo set di base STO-6G in modo da poter simulare l'esperimento e verificare che funzioni.
Passo 1: Mappare gli input classici su un problema quantistico
Prima di tutto, specifichiamo la molecola e le sue proprietà.
# Specify molecule properties
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="sto-6g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Compute exact energy using FCI
reference_energy = cas.run().e_tot
print(f"norb = {norb}")
print(f"nelec = {nelec}")
converged SCF energy = -108.464957764796
CASCI E = -108.595987350986 E(CI) = -32.4115475088426 S^2 = 0.0000000
norb = 8
nelec = (5, 5)
Prima di costruire il circuito ansatz LUCJ, eseguiamo un calcolo CCSD nella seguente cella di codice. Le ampiezze e di questo calcolo saranno usate per inizializzare i parametri dell'ansatz.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -108.5933309085008 E_corr = -0.1283731437052354
Ora usiamo ffsim per creare il circuito ansatz. Poiché la nostra molecola ha uno stato di Hartree-Fock a guscio chiuso, usiamo la variante spin-bilanciata dell'ansatz UCJ, UCJOpSpinBalanced. Impostiamo optimize=True nel metodo from_t_amplitudes per abilitare la fattorizzazione doppia "compressa" delle ampiezze (vedi The local unitary cluster Jastrow (LUCJ) ansatz nella documentazione di ffsim per i dettagli).
Poiché l'ansatz LUCJ si adatta alla connettività disponibile della QPU, dobbiamo inizializzare il backend QPU prima di creare l'ansatz. Per ora, creeremo un backend generico con una mappa di accoppiamento heavy-hex e un insieme di gate a cui l'ansatz LUCJ si decompone naturalmente. Poi useremo ffsim.qiskit.generate_lucj_pass_manager per creare un pass manager specializzato per transpilare l'ansatz LUCJ al backend dato secondo il layout a "zig-zag" descritto nella sezione di background sull'ansatz LUCJ. Questa funzione usa un'euristica di punteggio per minimizzare gli errori associati al layout selezionato, il che è importante se il tuo backend è una QPU reale, o un simulatore con un modello di rumore. Oltre a restituire il pass manager, questa funzione restituisce anche le coppie di accoppiamento alfa-beta che possono essere implementate sull'hardware. Se non tutte le coppie possono essere implementate, viene emesso un avviso.
import warnings
from qiskit.transpiler import CouplingMap
warnings.formatwarning = lambda msg, *args, **kwargs: f"Warning: {msg}\n"
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
coupling_map = CouplingMap.from_heavy_hex(3)
backend = GenericBackendV2(
coupling_map.size(),
coupling_map=coupling_map,
basis_gates=["cp", "xx_plus_yy", "p", "x", "swap"],
)
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Passo 2: Ottimizzare per l'esecuzione su hardware quantistico
Successivamente, ottimizziamo il circuito per un hardware target. Tipicamente, questo passaggio comporta l'inizializzazione del backend hardware e di un pass manager per quel backend. Tuttavia, poiché l'ansatz LUCJ è adattato alla connettività dell'hardware, abbiamo già eseguito queste operazioni nel passaggio precedente. Tutto ciò che resta da fare è eseguire il pass manager sul circuito per transpilarlo in un circuito ISA che può essere eseguito direttamente sulla QPU.
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
Gate counts: OrderedDict({'xx_plus_yy': 86, 'p': 16, 'measure': 16, 'cp': 15, 'x': 10, 'swap': 2, 'barrier': 1})
Passo 3: Eseguire usando le primitive Qiskit
Dopo aver ottimizzato il circuito per l'esecuzione hardware, siamo pronti ad eseguirlo sull'hardware target e raccogliere campioni per la stima dell'energia dello stato fondamentale. Poiché abbiamo un solo circuito, useremo la modalità di esecuzione Job di Qiskit Runtime ed eseguiremo il nostro circuito.
rng = np.random.default_rng()
sampler = StatevectorSampler(seed=rng)
job = sampler.run([isa_circuit], shots=100_000)
Warning: Trying to add QuantumRegister to a QuantumCircuit having a layout
primitive_result = job.result()
pub_result = primitive_result[0]
Passo 4: Post-elaborare e restituire il risultato nel formato classico desiderato
Una metrica utile per valutare la qualità dell'output della QPU è il numero di configurazioni valide restituite. Una configurazione valida ha il corretto numero di particelle e spin Z, il che significa che la metà destra del bitstring ha peso di Hamming uguale al numero di elettroni con spin up, e la metà sinistra ha peso di Hamming uguale al numero di elettroni con spin down. La seguente cella calcola la frazione di configurazioni campionate che sono valide.
def is_valid_bitstring(
bitstring: str, norb: int, nelec: tuple[int, int]
) -> bool:
n_alpha, n_beta = nelec
return (
len(bitstring) == 2 * norb
and bitstring[norb:].count("1") == n_alpha
and bitstring[:norb].count("1") == n_beta
)
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
Fraction of sampled configurations that are valid: 1.0
Tutti i bitstring sono validi perché stiamo campionando il circuito su un simulatore senza rumore. Quando si esegue su una QPU rumorosa, la frazione sarà inferiore a uno, ma si spera che sia maggiore della frazione che ci si aspetterebbe se i bitstring fossero campionati in modo uniforme a caso, che viene calcolata nella seguente cella.
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
Expected fraction of valid configurations from uniformly random bitstrings: 0.0478515625
Ora stimiamo l'energia dello stato fondamentale dell'Hamiltoniano usando la funzione diagonalize_fermionic_hamiltonian. Questa funzione esegue la procedura di recupero della configurazione auto-consistente per raffinare iterativamente i campioni quantistici rumorosi e migliorare la stima dell'energia. Passiamo una funzione callback per poter salvare i risultati intermedi per un'analisi successiva. Vedi la documentazione API per le spiegazioni degli argomenti di diagonalize_fermionic_hamiltonian.
Qui usiamo l'argomento initial_occupancies di diagonalize_fermionic_hamiltonian per specificare la configurazione Hartree-Fock come stima iniziale delle occupazioni degli orbitali nello stato fondamentale. Questo approccio è ragionevole per sistemi in cui lo stato fondamentale ha un supporto significativo sulla configurazione Hartree-Fock, ma potrebbe non essere appropriato in altre situazioni, anche se metodi computazionali più avanzati potrebbero fornire stime iniziali migliori in quei casi. Specificare initial_occupancies consente anche al recupero della configurazione di funzionare anche se non sono state campionate configurazioni valide, come può accadere quando si campiona un grande circuito su una QPU rumorosa. Senza questo argomento, il recupero della configurazione fallirebbe e genererebbe un errore se non venissero fornite configurazioni valide.
from functools import partial
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
solve_sci_batch,
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
Iteration 1
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Iteration 2
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Final energy: -108.59275573641656
Final energy error: 0.0032316145694579745
Visualizzare i risultati
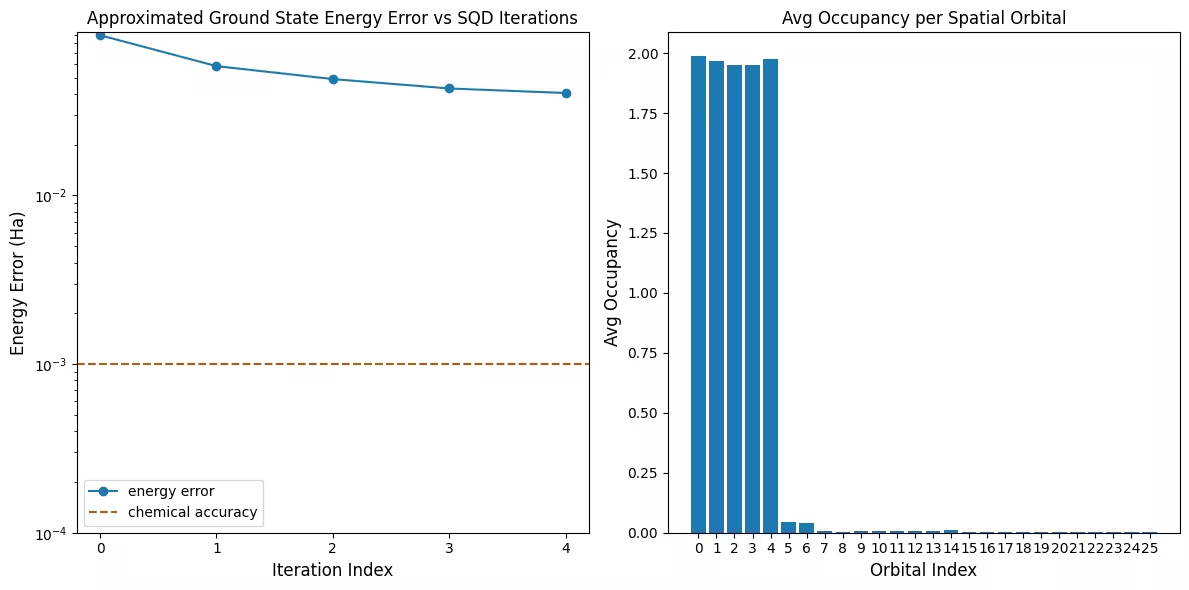
Il primo grafico mostra che in questa simulazione siamo già entro 1 mH dalla risposta esatta dopo la prima iterazione (l'accuratezza chimica è tipicamente accettata come 1 kcal/mol 1.6 mH). Questo è un sistema piccolo, però, e poiché i campioni sono privi di rumore, il recupero della configurazione non è necessario. Su un sistema più grande eseguito su una QPU rumorosa, potrebbero essere necessarie più iterazioni di recupero della configurazione, e l'accuratezza finale potrebbe essere peggiore. In generale, l'energia può essere migliorata consentendo più iterazioni di recupero della configurazione o aumentando il numero di campioni per batch.
Il secondo grafico mostra l'occupazione media di ciascun orbitale spaziale dopo l'iterazione finale. Possiamo vedere che sia gli elettroni con spin up che quelli con spin down occupano i primi cinque orbitali con alta probabilità nelle nostre soluzioni.
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()

Esempio su hardware reale su larga scala
Ora eseguiamo un esempio più grande su hardware quantistico reale. Qui, ricaveremo uno spazio attivo per la molecola di azoto dal set di base cc-pVDZ.
Passi 1-4
Qui mettiamo insieme tutti i passi in un unico flusso di lavoro su scala maggiore, che viene poi eseguito su hardware quantistico reale.
# ------------------------------ Step 1 ------------------------------
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="cc-pvdz",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Store reference energy from SCI calculation performed separately
reference_energy = -109.22802921665716
print(f"norb = {norb}")
print(f"nelec = {nelec}")
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=133
)
print(f"Using backend {backend.name}")
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
# ------------------------------ Step 2 ------------------------------
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
# ------------------------------ Step 3 ------------------------------
sampler = Sampler(mode=backend)
sampler.options.environment.job_tags = ["TUT_SQD"]
job = sampler.run([isa_circuit], shots=100_000)
primitive_result = job.result()
pub_result = primitive_result[0]
# ------------------------------ Step 4 ------------------------------
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the
# orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the
# sci_solver argument in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()
converged SCF energy = -108.929838385609
norb = 26
nelec = (5, 5)
E(CCSD) = -109.2177884185544 E_corr = -0.2879500329450045
Using backend ibm_boston
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20), (24, 24)].
Removing interaction (24, 24) from the end.
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20)].
Removing interaction (20, 20) from the end.
Gate counts: OrderedDict({'sx': 7039, 'rz': 6990, 'cz': 1858, 'x': 61, 'measure': 52, 'barrier': 1})
Fraction of sampled configurations that are valid: 0.02124
Expected fraction of valid configurations from uniformly random bitstrings: 9.607888706852918e-07
Iteration 1
Subsample 0
Energy: -109.13889134249762
Subspace dimension: 120409
Subsample 1
Energy: -109.11785470455858
Subspace dimension: 110889
Subsample 2
Energy: -109.13234360554011
Subspace dimension: 130321
Iteration 2
Subsample 0
Energy: -109.16392179579177
Subspace dimension: 223729
Subsample 1
Energy: -109.16281938332986
Subspace dimension: 223729
Subsample 2
Energy: -109.16955816711932
Subspace dimension: 233289
Iteration 3
Subsample 0
Energy: -109.17905772999075
Subspace dimension: 324900
Subsample 1
Energy: -109.17532445048462
Subspace dimension: 357604
Subsample 2
Energy: -109.1733168689756
Subspace dimension: 348100
Iteration 4
Subsample 0
Energy: -109.18437778820451
Subspace dimension: 474721
Subsample 1
Energy: -109.18450164209159
Subspace dimension: 476100
Subsample 2
Energy: -109.18493571190754
Subspace dimension: 487204
Iteration 5
Subsample 0
Energy: -109.18616522497996
Subspace dimension: 622521
Subsample 1
Energy: -109.18652868888333
Subspace dimension: 644809
Subsample 2
Energy: -109.18753326484406
Subspace dimension: 585225
Final energy: -109.18753326484406
Final energy error: 0.040495951813099396
Passi successivi
Se hai trovato questo lavoro interessante, potresti essere interessato al seguente materiale:
- Diagonalizzazione quantistica di Krylov basata su campioni di un modello reticolare fermionico - un tutorial correlato che usa circuiti di evoluzione temporale invece di un ansatz variazionale
- Scalare i flussi di lavoro SQD di chimica con il solver Dice - una pagina che mostra come usare il software Dice più efficiente per la diagonalizzazione
- Documentazione API del componente aggiuntivo SQD - riferimento per la funzione
diagonalize_fermionic_hamiltonian - Chemistry beyond the scale of exact diagonalization on a quantum-centric supercomputer - l'articolo su cui si basa questo tutorial