Diagonalizzazione quantistica di Krylov basata su campioni di un modello reticolare fermionico
Stima di utilizzo: Nove secondi su un processore Heron r2 (NOTA: Questa è solo una stima. Il tempo di esecuzione potrebbe variare.)
Risultati di apprendimento
Dopo aver completato questo tutorial, dovresti capire:
- Come usare il componente aggiuntivo SQD per Qiskit per approssimare l'energia dello stato fondamentale di un modello reticolare usando bitstring campionati da un'unità di elaborazione quantistica (QPU).
- Come usare ffsim per costruire circuiti di evoluzione temporale per la simulazione fermionica.
- Come combinare campioni da più circuiti per il post-processing con l'algoritmo di diagonalizzazione di Krylov basata su campioni (SKQD).
Prerequisiti
Suggeriamo di avere familiarità con i seguenti argomenti prima di affrontare questo tutorial:
- Diagonalizzazione quantistica basata su campioni di un Hamiltoniano di chimica
- Diagonalizzazione quantistica di Krylov degli Hamiltoniani reticolari
- Primitive Qiskit
Contesto
Questo tutorial mostra come utilizzare la diagonalizzazione quantistica basata su campioni (SQD) per stimare l'energia dello stato fondamentale di un modello reticolare fermionico. Nello specifico, studiamo il modello di Anderson a singola impurità unidimensionale (SIAM), utilizzato per descrivere impurità magnetiche incorporate nei metalli.
Questo tutorial segue un flusso di lavoro simile al tutorial correlato Diagonalizzazione quantistica basata su campioni di un Hamiltoniano di chimica. Tuttavia, una differenza chiave risiede nel modo in cui vengono costruiti i circuiti quantistici. L'altro tutorial utilizza un ansatz variazionale euristico, appropriato per Hamiltoniani di chimica con potenzialmente milioni di termini di interazione. Questo tutorial invece usa circuiti che approssimano l'evoluzione temporale dell'Hamiltoniano. Tali circuiti possono essere profondi, il che rende questo approccio migliore per applicazioni a modelli reticolari. I vettori di stato preparati da questi circuiti formano la base per un sottospazio di Krylov, e di conseguenza l'algoritmo converge in modo dimostrabile ed efficiente allo stato fondamentale, sotto opportune ipotesi.
L'approccio usato in questo tutorial può essere visto come una combinazione delle tecniche usate in SQD e nella diagonalizzazione quantistica di Krylov (KQD). L'approccio combinato è talvolta indicato come diagonalizzazione quantistica di Krylov basata su campioni (SQKD). Consulta Diagonalizzazione quantistica di Krylov degli Hamiltoniani reticolari per un tutorial sul metodo KQD.
Questo tutorial si basa sul lavoro "Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization", a cui puoi fare riferimento per maggiori dettagli.
Modello di Anderson a singola impurità (SIAM)
L'Hamiltoniano SIAM unidimensionale è una somma di tre termini:
dove
Qui, sono gli operatori fermionici di creazione/annichilazione per il sito del bagno con spin , sono gli operatori di creazione/annichilazione per il modo di impurità, e . , e sono numeri reali che descrivono le interazioni di hopping, on-site e ibridazione, mentre è un numero reale che specifica il potenziale chimico.
Nota che l'Hamiltoniano è un'istanza specifica dell'Hamiltoniano generico di elettroni interagenti,
dove consiste nei termini a un corpo, quadratici negli operatori fermionici di creazione e annichilazione, e consiste nei termini a due corpi, quartici. Per il SIAM,
e contiene il resto dei termini dell'Hamiltoniano. Per rappresentare l'Hamiltoniano in modo programmatico, memorizziamo la matrice e il tensore .
Basi di posizione e di impulso
A causa della simmetria traslazionale approssimativa in , non ci aspettiamo che lo stato fondamentale sia sparso nella base di posizione (la base orbitale in cui l'Hamiltoniano è specificato sopra). Le prestazioni di SQD sono garantite solo se lo stato fondamentale è sparso, cioè ha peso significativo su un numero ridotto di stati della base computazionale. Per migliorare la sparsità dello stato fondamentale, eseguiamo la simulazione nella base orbitale in cui è diagonale. Chiamiamo questa base la base degli impulsi. Poiché è un Hamiltoniano fermionico quadratico, può essere diagonalizzato efficientemente tramite una rotazione orbitale.
Evoluzione temporale approssimativa tramite l'Hamiltoniano
Per approssimare l'evoluzione temporale tramite l'Hamiltoniano, usiamo una decomposizione di Trotter-Suzuki del secondo ordine,
Sotto la trasformazione di Jordan-Wigner, l'evoluzione temporale tramite corrisponde a un singolo gate CPhase tra gli orbitali spin-up e spin-down al sito dell'impurità. Poiché è un Hamiltoniano fermionico quadratico, l'evoluzione temporale tramite corrisponde a una rotazione orbitale.
Gli stati della base di Krylov , dove è la dimensione del sottospazio di Krylov, sono formati dall'applicazione ripetuta di un singolo passo di Trotter, quindi
Nel seguente flusso di lavoro basato su SQD, campioneremo da questo insieme di circuiti e post-processeremo l'insieme combinato di bitstring con SQD. Questo approccio contrasta con quello usato nel tutorial correlato Diagonalizzazione quantistica basata su campioni di un Hamiltoniano di chimica, dove i campioni venivano estratti da un singolo circuito variazionale euristico.
Requisiti
Prima di iniziare questo tutorial, assicurati di avere installato quanto segue:
- Qiskit SDK v1.0 o successivo, con supporto per la visualizzazione
- Qiskit Runtime v0.22 o successivo (
pip install qiskit-ibm-runtime) - SQD Qiskit addon v0.11 o successivo (
pip install qiskit-addon-sqd) - ffsim v0.0.72 o successivo (
pip install ffsim)
Esempio su simulatore su piccola scala
Passo 1: Mappare il problema su un circuito quantistico
Per prima cosa, generiamo l'Hamiltoniano SIAM nella base di posizione. L'Hamiltoniano è rappresentato dalla matrice e dal tensore . Poi lo ruotiamo nella base degli impulsi. Nella base di posizione, poniamo l'impurità al primo sito. Tuttavia, quando ruotiamo nella base degli impulsi, spostiamo l'impurità in un sito centrale per facilitare le interazioni con altri orbitali.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime scipy
import numpy as np
import pyscf.fci
def siam_hamiltonian(
norb: int,
hopping: float,
onsite: float,
hybridization: float,
chemical_potential: float,
) -> tuple[np.ndarray, np.ndarray]:
"""Hamiltonian for the single-impurity Anderson model."""
# Place the impurity on the first site
impurity_orb = 0
# One body matrix elements in the "position" basis
h1e = np.zeros((norb, norb))
np.fill_diagonal(h1e[:, 1:], -hopping)
np.fill_diagonal(h1e[1:, :], -hopping)
h1e[impurity_orb, impurity_orb + 1] = -hybridization
h1e[impurity_orb + 1, impurity_orb] = -hybridization
h1e[impurity_orb, impurity_orb] = chemical_potential
# Two body matrix elements in the "position" basis
h2e = np.zeros((norb, norb, norb, norb))
h2e[impurity_orb, impurity_orb, impurity_orb, impurity_orb] = onsite
return h1e, h2e
def momentum_basis(norb: int) -> np.ndarray:
"""Get the orbital rotation to change from the position to the momentum basis."""
n_bath = norb - 1
# Orbital rotation that diagonalizes the bath (non-interacting system)
hopping_matrix = np.zeros((n_bath, n_bath))
np.fill_diagonal(hopping_matrix[:, 1:], -1)
np.fill_diagonal(hopping_matrix[1:, :], -1)
_, vecs = np.linalg.eigh(hopping_matrix)
# Expand to include impurity
orbital_rotation = np.zeros((norb, norb))
# Impurity is on the first site
orbital_rotation[0, 0] = 1
orbital_rotation[1:, 1:] = vecs
# Move the impurity to the center
new_index = n_bath // 2
perm = np.r_[1 : (new_index + 1), 0, (new_index + 1) : norb]
orbital_rotation = orbital_rotation[:, perm]
return orbital_rotation
def rotated(
h1e: np.ndarray, h2e: np.ndarray, orbital_rotation: np.ndarray
) -> tuple[np.ndarray, np.ndarray]:
"""Rotate the orbital basis of a Hamiltonian."""
h1e_rotated = np.einsum(
"ab,Aa,Bb->AB",
h1e,
orbital_rotation,
orbital_rotation.conj(),
optimize="greedy",
)
h2e_rotated = np.einsum(
"abcd,Aa,Bb,Cc,Dd->ABCD",
h2e,
orbital_rotation,
orbital_rotation.conj(),
orbital_rotation,
orbital_rotation.conj(),
optimize="greedy",
)
return h1e_rotated, h2e_rotated
# Total number of spatial orbitals, including the bath sites and the impurity
# This should be an even number
norb = 8
# System is half-filled
nelec = (norb // 2, norb // 2)
# One orbital is the impurity, the rest are bath sites
n_bath = norb - 1
# Hamiltonian parameters
hybridization = 1.0
hopping = 1.0
onsite = 10.0
chemical_potential = -0.5 * onsite
# Generate Hamiltonian in position basis
h1e, h2e = siam_hamiltonian(
norb=norb,
hopping=hopping,
onsite=onsite,
hybridization=hybridization,
chemical_potential=chemical_potential,
)
# Rotate to momentum basis
orbital_rotation = momentum_basis(norb)
h1e_momentum, h2e_momentum = rotated(h1e, h2e, orbital_rotation.T.conj())
# In the momentum basis, the impurity is placed in the center
impurity_index = n_bath // 2
# Use PySCF to compute the exact ground state energy
reference_energy, _ = pyscf.fci.direct_spin1.kernel(h1e, h2e, norb, nelec)
Successivamente, generiamo i circuiti per produrre gli stati della base di Krylov. Per ogni specie di spin, lo stato iniziale è dato dalla sovrapposizione di tutte le possibili eccitazioni dei tre elettroni più vicini al livello di Fermi nei 4 modi vuoti più vicini, a partire dallo stato , e realizzata dall'applicazione di sette gate XXPlusYYGate. Gli stati evoluti nel tempo sono prodotti da applicazioni successive di un passo di Trotter del secondo ordine.
Per una descrizione più dettagliata di questo modello e di come vengono progettati i circuiti, consulta "Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization".
from typing import Sequence
import ffsim
import scipy
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.circuit import CircuitInstruction, Qubit
from qiskit.circuit.library import CPhaseGate, XGate, XXPlusYYGate
def prepare_initial_state(qubits: Sequence[Qubit], norb: int, nocc: int):
"""Prepare initial state."""
assert norb >= 8
x_gate = XGate()
rot = XXPlusYYGate(0.5 * np.pi, -0.5 * np.pi)
for i in range(nocc):
yield CircuitInstruction(x_gate, [qubits[i]])
yield CircuitInstruction(x_gate, [qubits[norb + i]])
for i in range(3):
for j in range(nocc - i - 1, nocc + i, 2):
yield CircuitInstruction(rot, [qubits[j], qubits[j + 1]])
yield CircuitInstruction(
rot, [qubits[norb + j], qubits[norb + j + 1]]
)
yield CircuitInstruction(rot, [qubits[j + 1], qubits[j + 2]])
yield CircuitInstruction(
rot, [qubits[norb + j + 1], qubits[norb + j + 2]]
)
def trotter_step(
qubits: Sequence[Qubit],
time_step: float,
one_body_evolution: np.ndarray,
h2e: np.ndarray,
impurity_index: int,
norb: int,
):
"""A Trotter step."""
# Assume the two-body interaction is just the on-site interaction of the impurity
onsite = h2e[
impurity_index, impurity_index, impurity_index, impurity_index
]
# Two-body evolution for half the time
yield CircuitInstruction(
CPhaseGate(-0.5 * time_step * onsite),
[qubits[impurity_index], qubits[norb + impurity_index]],
)
# One-body evolution for the full time
yield CircuitInstruction(
ffsim.qiskit.OrbitalRotationJW(norb, one_body_evolution), qubits
)
# Two-body evolution for half the time
yield CircuitInstruction(
CPhaseGate(-0.5 * time_step * onsite),
[qubits[impurity_index], qubits[norb + impurity_index]],
)
# Time step
time_step = 0.2
# Number of Krylov basis states
krylov_dim = 8
# Initialize circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# Generate initial state
for instruction in prepare_initial_state(qubits, norb=norb, nocc=norb // 2):
circuit.append(instruction)
circuit.measure_all()
# Create list of circuits, starting with the initial state circuit
circuits = [circuit.copy()]
# Add time evolution circuits to the list
one_body_evolution = scipy.linalg.expm(-1j * time_step * h1e_momentum)
for i in range(krylov_dim - 1):
# Remove measurements
circuit.remove_final_measurements()
# Append another Trotter step
for instruction in trotter_step(
qubits,
time_step,
one_body_evolution,
h2e_momentum,
impurity_index,
norb,
):
circuit.append(instruction)
# Measure qubits
circuit.measure_all()
# Add a copy of the circuit to the list
circuits.append(circuit.copy())
circuits[0].draw("mpl", scale=0.4, fold=-1)
circuits[-1].draw("mpl", scale=0.4, fold=-1)
Passo 2: Ottimizzare il problema per l'esecuzione su hardware quantistico
Ora ottimizziamo il circuito per un hardware target. Per ora creeremo un backend generico con un numero specificato di qubit e un insieme di gate a cui i circuiti di evoluzione temporale si decompongono naturalmente.
from qiskit.providers.fake_provider import GenericBackendV2
backend = GenericBackendV2(
2 * norb, basis_gates=["cp", "xx_plus_yy", "p", "x"]
)
Ora usiamo Qiskit per trasporre i circuiti verso il backend target.
from qiskit.transpiler import generate_preset_pass_manager
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend
)
isa_circuits = pass_manager.run(circuits)
Passo 3: Eseguire con le primitive Qiskit
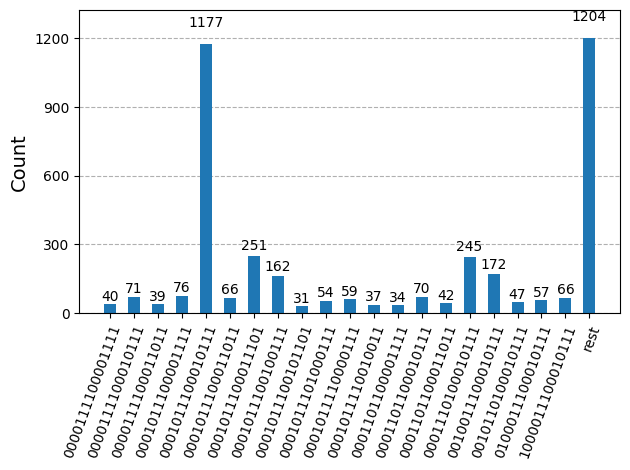
Dopo aver ottimizzato i circuiti per l'esecuzione su hardware, siamo pronti per eseguirli sull'hardware target e raccogliere campioni per la stima dell'energia dello stato fondamentale. Dopo aver usato la primitiva Sampler per campionare bitstring da ciascun circuito, combiniamo tutti i risultati in un singolo dizionario di conteggi e tracciamo i 20 bitstring campionati più frequentemente.
from qiskit.visualization import plot_histogram
from qiskit.primitives import StatevectorSampler
# Sample from the circuits
sampler = StatevectorSampler()
job = sampler.run(isa_circuits, shots=500)
from qiskit.primitives import BitArray
# Combine the shots from the individual Trotter circuits
bit_array = BitArray.concatenate_shots(
[result.data.meas for result in job.result()]
)
plot_histogram(bit_array.get_counts(), number_to_keep=20)
Passo 4: Post-elaborare e restituire il risultato nel formato classico desiderato
Ora eseguiamo l'algoritmo SQD usando la funzione diagonalize_fermionic_hamiltonian. Consulta la documentazione API per le spiegazioni degli argomenti di questa funzione.
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
rng = np.random.default_rng(24)
result = diagonalize_fermionic_hamiltonian(
h1e_momentum,
h2e_momentum,
bit_array,
samples_per_batch=100,
norb=norb,
nelec=nelec,
num_batches=3,
max_iterations=5,
symmetrize_spin=True,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -13.4222953188441
Subspace dimension: 529
Subsample 1
Energy: -13.42237556285828
Subspace dimension: 784
Subsample 2
Energy: -13.422045397387413
Subspace dimension: 529
Iteration 2
Subsample 0
Energy: -13.422379583305478
Subspace dimension: 900
Subsample 1
Energy: -13.422376197704326
Subspace dimension: 841
Subsample 2
Energy: -13.422421162849295
Subspace dimension: 1089
Iteration 3
Subsample 0
Energy: -13.422421164670345
Subspace dimension: 1156
Subsample 1
Energy: -13.422421492737689
Subspace dimension: 1156
Subsample 2
Energy: -13.422421205869572
Subspace dimension: 1156
Iteration 4
Subsample 0
Energy: -13.422421494558726
Subspace dimension: 1225
Subsample 1
Energy: -13.422421492737689
Subspace dimension: 1156
Subsample 2
Energy: -13.422421492737689
Subspace dimension: 1156
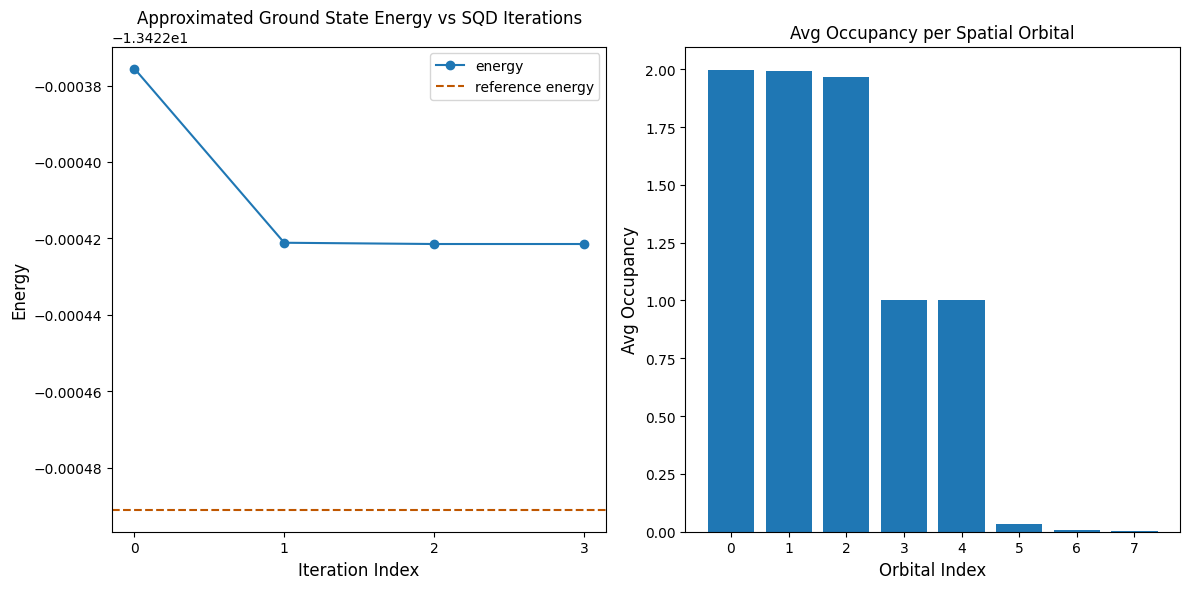
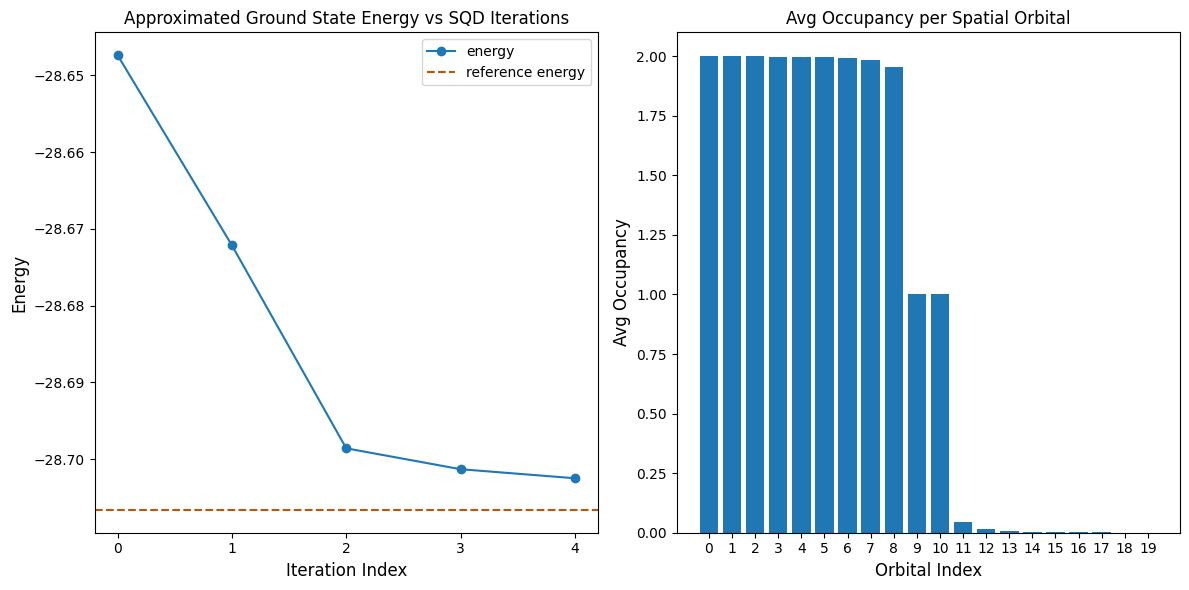
La seguente cella di codice traccia i risultati. Il primo grafico mostra l'energia calcolata in funzione del numero di iterazioni di recupero della configurazione, e il secondo mostra l'occupazione media di ciascun orbitale spaziale dopo l'ultima iterazione. Poiché si tratta di un problema così piccolo, la prima iterazione ci porta già molto vicini all'energia esatta (nota la scala dell'asse y).
import matplotlib.pyplot as plt
min_es = [
min(result, key=lambda res: res.energy).energy
for result in result_history
]
min_id, min_e = min(enumerate(min_es), key=lambda x: x[1])
# Data for energies plot
x1 = range(len(result_history))
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, min_es, label="energy", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].axhline(
y=reference_energy,
color="#BF5700",
linestyle="--",
label="reference energy",
)
axs[0].set_title("Approximated Ground State Energy vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Reference energy: {reference_energy:.5f}")
print(f"SQD energy: {min_e:.5f}")
print(f"Absolute error: {abs(min_e - reference_energy):.5f}")
plt.tight_layout()
plt.show()
Reference energy: -13.42249
SQD energy: -13.42242
Absolute error: 0.00007
Verifica dell'energia
L'energia restituita da SQD è garantita essere un limite superiore alla vera energia dello stato fondamentale. Il valore dell'energia può essere verificato perché SQD restituisce anche i coefficienti del vettore di stato che approssima lo stato fondamentale. Puoi calcolare l'energia dal vettore di stato usando le sue matrici di densità ridotta a una e due particelle, come dimostrato nella seguente cella di codice.
rdm1 = result.sci_state.rdm(rank=1, spin_summed=True)
rdm2 = result.sci_state.rdm(rank=2, spin_summed=True)
energy = np.sum(h1e_momentum * rdm1) + 0.5 * np.sum(h2e_momentum * rdm2)
print(f"Recomputed energy: {energy:.5f}")
Recomputed energy: -13.42242
Esempio su hardware su larga scala
Ora eseguiamo un esempio più grande su una QPU reale. Per l'energia di riferimento, usiamo i risultati di un calcolo DMRG eseguito separatamente.
from qiskit_ibm_runtime import SamplerV2 as Sampler
from qiskit_ibm_runtime import QiskitRuntimeService
# Model parameters
norb = 20
nelec = (norb // 2, norb // 2)
n_bath = norb - 1
hybridization = 1.0
hopping = 1.0
onsite = 10.0
chemical_potential = -0.5 * onsite
# Generate Hamiltonian and orbital rotation
h1e, h2e = siam_hamiltonian(
norb=norb,
hopping=hopping,
onsite=onsite,
hybridization=hybridization,
chemical_potential=chemical_potential,
)
orbital_rotation = momentum_basis(norb)
h1e_momentum, h2e_momentum = rotated(h1e, h2e, orbital_rotation.T.conj())
impurity_index = n_bath // 2
# Set reference energy to DMRG value computed separately
reference_energy = -28.70659686
# Algorithm parameters
time_step = 0.2
krylov_dim = 8
# Construct circuits
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
for instruction in prepare_initial_state(qubits, norb=norb, nocc=norb // 2):
circuit.append(instruction)
circuit.measure_all()
circuits = [circuit.copy()]
one_body_evolution = scipy.linalg.expm(-1j * time_step * h1e_momentum)
for i in range(krylov_dim - 1):
circuit.remove_final_measurements()
for instruction in trotter_step(
qubits,
time_step,
one_body_evolution,
h2e_momentum,
impurity_index,
norb,
):
circuit.append(instruction)
circuit.measure_all()
circuits.append(circuit.copy())
# Initialize hardware backend
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=127
)
print(f"Using backend {backend.name}")
# Transpile to backend
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend
)
isa_circuits = pass_manager.run(circuits)
# Sample from the circuits
sampler = Sampler(backend)
sampler.options.environment.job_tags = ["TUT_SKQD"]
job = sampler.run(isa_circuits, shots=500)
# Combine the shots from the individual Trotter circuits
bit_array = BitArray.concatenate_shots(
[result.data.meas for result in job.result()]
)
# Run configuration recovery and diagonalization
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
rng = np.random.default_rng(24)
result = diagonalize_fermionic_hamiltonian(
h1e_momentum,
h2e_momentum,
bit_array,
samples_per_batch=100,
norb=norb,
nelec=nelec,
num_batches=3,
max_iterations=5,
symmetrize_spin=True,
callback=callback,
seed=rng,
)
# Plot results
min_es = [
min(result, key=lambda res: res.energy).energy
for result in result_history
]
min_id, min_e = min(enumerate(min_es), key=lambda x: x[1])
x1 = range(len(result_history))
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
axs[0].plot(x1, min_es, label="energy", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].axhline(
y=reference_energy,
color="#BF5700",
linestyle="--",
label="reference energy",
)
axs[0].set_title("Approximated Ground State Energy vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy", fontdict={"fontsize": 12})
axs[0].legend()
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Reference energy: {reference_energy:.5f}")
print(f"SQD energy: {min_e:.5f}")
print(f"Absolute error: {abs(min_e - reference_energy):.5f}")
plt.tight_layout()
plt.show()
Using backend ibm_boston
Iteration 1
Subsample 0
Energy: -28.63965951544449
Subspace dimension: 9801
Subsample 1
Energy: -28.625588929202006
Subspace dimension: 9409
Subsample 2
Energy: -28.647371834135498
Subspace dimension: 8281
Iteration 2
Subsample 0
Energy: -28.67213260849567
Subspace dimension: 29584
Subsample 1
Energy: -28.670340686158816
Subspace dimension: 27225
Subsample 2
Energy: -28.669976379525988
Subspace dimension: 31329
Iteration 3
Subsample 0
Energy: -28.68622875601382
Subspace dimension: 36100
Subsample 1
Energy: -28.698569623143126
Subspace dimension: 34225
Subsample 2
Energy: -28.694848533971882
Subspace dimension: 33856
Iteration 4
Subsample 0
Energy: -28.69883392844593
Subspace dimension: 42025
Subsample 1
Energy: -28.701289495200996
Subspace dimension: 38025
Subsample 2
Energy: -28.699319594978245
Subspace dimension: 45369
Iteration 5
Subsample 0
Energy: -28.701936886834154
Subspace dimension: 51076
Subsample 1
Energy: -28.702468711812013
Subspace dimension: 53824
Subsample 2
Energy: -28.702298147575938
Subspace dimension: 52900
Reference energy: -28.70660
SQD energy: -28.70247
Absolute error: 0.00413
Passi successivi
Se hai trovato questo lavoro interessante, potresti essere interessato ai seguenti materiali:
- Diagonalizzazione quantistica basata su campioni di un Hamiltoniano di chimica - un tutorial correlato che usa un ansatz variazionale euristico invece di circuiti di Trotter
- Diagonalizzazione quantistica di Krylov degli Hamiltoniani reticolari - un tutorial sul metodo KQD
- Documentazione API dell'addon SQD - riferimento per la funzione
diagonalize_fermionic_hamiltonian - Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization - il paper su cui si basa questo tutorial