Diagonalizzazione quantistica di Krylov
In questa lezione sulla diagonalizzazione Krylov quantistica (KQD) risponderemo alle seguenti domande:
- Cos'è il metodo Krylov, in generale?
- Perché il metodo Krylov funziona e in quali condizioni?
- Che ruolo svolge il calcolo quantistico?
La parte quantistica dei calcoli si basa in gran parte sul lavoro del Rif. [1].
Il video seguente fornisce una panoramica dei metodi Krylov nel calcolo classico, ne motiva l'utilizzo e spiega come il calcolo quantistico possa svolgere un ruolo in quel flusso di lavoro. Il testo successivo offre maggiori dettagli e implementa un metodo Krylov sia in modo classico che su un computer quantistico.
1. Introduzione ai metodi Krylov
Un metodo per sottospazi di Krylov può fare riferimento a uno qualsiasi dei vari metodi costruiti attorno a quello che viene chiamato il sottospazio di Krylov. Una rassegna completa di questi metodi va oltre lo scopo di questa lezione, ma i Rif. [2-4] possono tutti fornire un background sostanzialmente più approfondito. Qui ci concentreremo su cosa sia un sottospazio di Krylov, come e perché sia utile nella risoluzione di problemi agli autovalori, e infine come possa essere implementato su un computer quantistico.
Definizione: Data una matrice simmetrica e semidefinita positiva , lo spazio di Krylov di ordine è lo spazio generato dai vettori ottenuti moltiplicando potenze crescenti della matrice , fino a , per un vettore di riferimento .
Sebbene i vettori sopra riportati generino quello che chiamiamo sottospazio di Krylov, non c'è ragione di pensare che siano ortogonali. Si usa spesso un processo di ortonormalizzazione iterativa simile all'ortonormalizzazione di Gram-Schmidt. Qui il processo è leggermente diverso, poiché ogni nuovo vettore viene reso ortogonale agli altri nel momento in cui viene generato. In questo contesto viene chiamato iterazione di Arnoldi. Partendo dal vettore iniziale , si genera il vettore successivo e poi si assicura che questo secondo vettore sia ortogonale al primo sottraendo la sua proiezione su . Ovvero
È ora facile vedere che poiché
Facciamo lo stesso per il vettore successivo, assicurandoci che sia ortogonale a entrambi i precedenti:
Se ripetiamo questo processo per tutti gli vettori, abbiamo una base ortonormale completa per uno spazio di Krylov. Si noti che il processo di ortonormalizzazione qui darà zero non appena , poiché vettori ortogonali necessariamente generano l'intero spazio. Il processo darà zero anche se un vettore è un autovettore di , poiché tutti i vettori successivi saranno multipli di quel vettore.
1.1 Un esempio semplice: Krylov a mano
Proviamo a generare un sottospazio di Krylov su una matrice di dimensioni banalmente piccole, in modo da poter osservare il processo passo per passo. Partiamo da una matrice iniziale di nostro interesse:
Per questo piccolo esempio possiamo determinare gli autovettori e gli autovalori facilmente anche a mano. Mostriamo qui la soluzione numerica.
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-ibm-runtime scipy sympy
# One might use linalg.eigh here, but later matrices may not be Hermitian. So we use
# linalg.eig in this lesson.
import numpy as np
A = np.array([[4, -1, 0], [-1, 4, -1], [0, -1, 4]])
eigenvalues, eigenvectors = np.linalg.eig(A)
print("The eigenvalues are ", eigenvalues)
print("The eigenvectors are ", eigenvectors)
The eigenvalues are [2.58578644 4. 5.41421356]
The eigenvectors are [[ 5.00000000e-01 -7.07106781e-01 5.00000000e-01]
[ 7.07106781e-01 1.37464400e-16 -7.07106781e-01]
[ 5.00000000e-01 7.07106781e-01 5.00000000e-01]]
Li annotiamo qui per un confronto successivo:
Vogliamo studiare come questo processo funziona (o fallisce) man mano che aumentiamo la dimensione del nostro sottospazio di Krylov, . A questo scopo, applicheremo questo processo:
- Generare un sottospazio dello spazio vettoriale completo partendo da un vettore scelto casualmente (chiamiamolo se è già normalizzato, come sopra).
- Proiettare la matrice completa su quel sottospazio e trovare gli autovalori di quella matrice proiettata .
- Aumentare la dimensione del sottospazio generando ulteriori vettori, assicurandosi che siano ortonormali, usando un processo simile all'ortonormalizzazione di Gram-Schmidt.
- Proiettare sul sottospazio più grande e trovare gli autovalori della matrice risultante, .
- Ripetere fino a quando gli autovalori convergono (oppure, in questo caso giocattolo, fino a quando si sono generati vettori che generano l'intero spazio vettoriale della matrice originale ).
Un'implementazione normale del metodo Krylov non avrebbe bisogno di risolvere il problema agli autovalori per la matrice proiettata su ogni sottospazio di Krylov nel momento in cui viene costruito. Si potrebbe costruire il sottospazio della dimensione desiderata, proiettare la matrice su quel sottospazio e diagonalizzare la matrice proiettata. La proiezione e la diagonalizzazione ad ogni dimensione del sottospazio vengono fatte solo per verificare la convergenza.
Dimensione :
Scegliamo un vettore casuale, ad esempio
Se non è già normalizzato, normalizziamolo.
Proiettiamo ora la nostra matrice sul sottospazio di questo singolo vettore:
Questa è la nostra proiezione della matrice sul nostro sottospazio di Krylov quando contiene un solo vettore, . L'autovalore di questa matrice è banalmente 4. Possiamo considerare questo come la nostra stima di ordine zero degli autovalori (in questo caso solo uno) di . Sebbene sia una stima approssimativa, è del giusto ordine di grandezza.
Dimensione :
Generiamo ora il vettore successivo nel nostro sottospazio attraverso l'operazione di sul vettore precedente:
Ora sottraiamo la proiezione di questo vettore sul vettore precedente per garantire l'ortogonalità.
Se non è già normalizzato, normalizziamolo. In questo caso il vettore era già normalizzato, quindi
Proiettiamo ora la nostra matrice A sul sottospazio di questi due vettori:
Rimane ancora il problema di determinare gli autovalori di questa matrice. Ma questa matrice è leggermente più piccola della matrice completa. In problemi che coinvolgono matrici molto grandi, lavorare con questo sottospazio più piccolo può essere molto vantaggioso.
Sebbene questa sia ancora una stima approssimativa, è migliore della stima di ordine zero. Porteremo avanti questo processo per un'altra iterazione, per assicurarci che il processo sia chiaro. Tuttavia, questo vanifica lo scopo del metodo, poiché nell'iterazione successiva finiremo per diagonalizzare una matrice 3x3, il che significa che non abbiamo risparmiato né tempo né potenza computazionale.
Dimensione :
Generiamo ora il vettore successivo nel nostro sottospazio attraverso l'operazione di A sul vettore precedente:
Ora sottraiamo la proiezione di questo vettore su entrambi i vettori precedenti per garantire l'ortogonalità.
Se non è già normalizzato, normalizziamolo. In questo caso il vettore era già normalizzato, quindi
Proiettiamo ora la nostra matrice sul sottospazio di questi vettori:
Determiniamo ora gli autovalori:
Questi autovalori sono esattamente gli autovalori della matrice originale . Questo deve essere il caso, poiché abbiamo espanso il nostro sottospazio di Krylov fino a coprire l'intero spazio vettoriale della matrice originale .
In questo esempio, il metodo Krylov potrebbe non sembrare particolarmente più semplice della diagonalizzazione diretta. Infatti, come vedremo nelle sezioni successive, il metodo Krylov è vantaggioso solo al di sopra di una certa dimensione della matrice; questo esempio è inteso ad aiutarci a risolvere problemi di autovalori/autovettori di matrici estremamente grandi.
Questo è l'unico esempio che mostreremo risolto "a mano", ma la sezione 2 qui sotto mostra esempi computazionali.
Chiarimento dei termini
Un malinteso comune è che esista un solo sottospazio di Krylov per un dato problema. Ma ovviamente, poiché ci sono molti vettori iniziali a cui la nostra matrice potrebbe essere applicata, esistono molti possibili sottospazi di Krylov. Useremo la frase "il sottospazio di Krylov" solo per fare riferimento a un sottospazio di Krylov specifico già definito per un esempio specifico. Per approcci generali alla risoluzione di problemi faremo riferimento a "un sottospazio di Krylov". Un'ultima precisazione è che è corretto parlare di "spazio di Krylov". Spesso lo si chiama "sottospazio di Krylov" a causa del suo utilizzo nel contesto della proiezione di matrici da uno spazio iniziale in un sottospazio. Coerentemente con quel contesto, ci riferiremo per lo più ad esso come a un sottospazio qui.
Verifica della comprensione
Spiega perché non è (a) utile, e (b) possibile estendere la dimensione del sottospazio di Krylov oltre la dimensione della matrice di interesse.
Answer
(a) Poiché ortonormalizziamo i vettori man mano che li produciamo, un insieme di tali vettori formerà una base completa, il che significa che una loro combinazione lineare può essere usata per creare qualsiasi vettore nello spazio.
(b) Il processo di ortonormalizzazione consiste nel sottrarre la proiezione di un nuovo vettore su tutti i vettori precedenti. Se tutti i vettori precedenti generano l'intero spazio vettoriale, sottrarre le proiezioni sull'intero sottospazio ci lascerà sempre con un vettore zero.
Supponi che un collega ricercatore stia dimostrando il metodo Krylov applicato a una piccola matrice giocattolo. C'è qualcosa di sbagliato nella scelta della matrice e del vettore iniziale ?
e
Answer
Il tuo collega ha scelto accidentalmente un autovettore come vettore iniziale. Applicare la matrice al vettore iniziale restituirà semplicemente lo stesso vettore, scalato dall'autovalore. Questo non genererà un sottospazio di dimensione crescente. Consiglia al tuo collega di scegliere un vettore iniziale diverso, assicurandosi che non sia un autovettore.
Applica il metodo Krylov alla matrice data, scegliendo un appropriato nuovo vettore iniziale. Scrivi le stime dell'autovalore minimo all'ordine 0 e all'ordine 1 del tuo sottospazio di Krylov.
Answer
Ci sono molte risposte possibili a seconda della scelta del vettore iniziale. Sceglieremo:
Per ottenere applichiamo una volta a , e poi rendiamo ortogonale a
All'ordine 0, la proiezione sul nostro sottospazio di Krylov è
All'ordine 1, la proiezione su questo sottospazio di Krylov è
Questo può essere fatto a mano, ma è più facilmente eseguibile usando numpy:
import numpy as np
vstar = np.array([[1/np.sqrt(3),1/np.sqrt(3),1/np.sqrt(3)],[-1/np.sqrt(6),np.sqrt(2/3),-1/np.sqrt(6)]]
)
A = np.array([[1, 1, 0],
[1, 1, 1],
[0, 1, 1]])
v = np.array([[1/np.sqrt(3),-1/np.sqrt(6)],[1/np.sqrt(3),np.sqrt(2/3)],[1/np.sqrt(3),-1/np.sqrt(6)]])
proj = vstar@A@v
print(proj)
eigenvalues, eigenvectors = np.linalg.eig(proj)
print("The eigenvalues are ", eigenvalues)
print("The eigenvectors are ", eigenvectors)
outputs:
[[ 2.33333333 0.47140452]
[ 0.47140452 -0.33333333]]
The eigenvalues are [ 2.41421356 -0.41421356]
The eigenvectors are [[ 0.98559856 -0.16910198]
[ 0.16910198 0.98559856]]
La stima dell'autovalore minimo è -0.414.
1.2 Tipi di metodi Krylov
I "metodi per sottospazi di Krylov" possono fare riferimento a qualsiasi delle varie tecniche iterative utilizzate per risolvere grandi sistemi lineari e problemi agli autovalori. Ciò che hanno tutti in comune è che costruiscono una soluzione approssimata da un sottospazio di Krylov
dove è la stima iniziale (vedi Rif. [5]). Differiscono nel modo in cui scelgono la migliore approssimazione da questo sottospazio, bilanciando fattori quali la velocità di convergenza, l'utilizzo della memoria e il costo computazionale complessivo. L'obiettivo di questa lezione è sfruttare il calcolo quantistico nel contesto dei metodi per sottospazi di Krylov; una discussione esaustiva di questi metodi va oltre il suo scopo. Le brevi definizioni qui sotto servono solo come contesto e includono alcuni riferimenti per approfondire ulteriormente questi metodi.
Il metodo del gradiente coniugato (CG): Questo metodo è usato per risolvere sistemi lineari simmetrici e definiti positivi[6]. Minimizza la norma-A dell'errore ad ogni iterazione, rendendolo particolarmente efficace per sistemi che derivano da EDP ellittiche discretizzate[7]. Utilizzeremo questo approccio nella prossima sezione per motivare perché un sottospazio di Krylov sarebbe un sottospazio efficace in cui cercare soluzioni migliori ai sistemi lineari.
Il metodo del residuo minimo generalizzato (GMRES): È progettato per risolvere sistemi lineari generali non simmetrici. Minimizza la norma del residuo su uno spazio di Krylov ad ogni iterazione, rendendolo robusto ma potenzialmente costoso in termini di memoria per sistemi di grandi dimensioni[7].
Il metodo del residuo minimo (MINRES): Questo metodo è usato per risolvere sistemi lineari simmetrici indefiniti. È simile al GMRES ma sfrutta la simmetria della matrice per ridurre il costo computazionale[8].
Altri approcci degni di nota includono il metodo di ortonormalizzazione completa (FOM), strettamente correlato al metodo di Arnoldi per i problemi agli autovalori, il metodo del gradiente bi-coniugato (BiCG) e il metodo di riduzione della dimensione indotta (IDR).
1.3 Perché il metodo per sottospazi di Krylov funziona
Qui motiveremo il fatto che il metodo per sottospazi di Krylov dovrebbe essere un modo efficiente per approssimare gli autovalori di una matrice tramite un raffinamento iterativo delle approssimazioni degli autovettori, attraverso la prospettiva della discesa più ripida. Sosterremo che, data una stima iniziale dello stato fondamentale, lo spazio delle correzioni successive a quella stima iniziale che produce la convergenza più rapida è un sottospazio di Krylov. Ci fermiamo prima di una dimostrazione rigorosa del comportamento di convergenza.
Supponiamo che la nostra matrice di interesse sia simmetrica e definita positiva. Questo rende il nostro argomento più rilevante per il metodo CG sopra. Non facciamo ipotesi sulla sparsità; né stiamo affermando che debba essere Hermitiana (cosa necessaria se è un'Hamiltoniana).
In genere vogliamo risolvere un problema della forma
Si potrebbe immaginare che dove è una costante, come in un problema agli autovalori. Ma la nostra formulazione del problema rimane più generale per ora.
Partiamo da un vettore che è una soluzione approssimata. Sebbene ci siano parallelismi tra questa stima e nella Sezione 1.1, non li sfruttiamo qui. La nostra stima ha un errore, che chiamiamo
Definiamo anche il residuo
Qui usiamo la maiuscola per distinguere il residuo dalla dimensione del nostro sottospazio di Krylov .

Vogliamo ora effettuare un passo di correzione della forma
che speriamo migliori la nostra approssimazione. Qui è un vettore ancora da determinare. Sia l'errore dopo che la correzione è stata effettuata. Allora

Siamo interessati al comportamento del nostro errore quando trasformato dalla nostra matrice. Calcoliamo quindi la norma- dell'errore. Ovvero
dove abbiamo usato la simmetria di e anche che Qui è una costante indipendente da . Come menzionato nella Sezione 1.2, la norma- dell'errore non è l'unica quantità che potremmo scegliere di minimizzare, ma è una buona scelta. Vogliamo vedere come questa quantità varia con la nostra scelta dei vettori di correzione Definiamo quindi la funzione impostando
è semplicemente l'errore come funzione della correzione misurata nella norma-. Di conseguenza, vogliamo scegliere in modo che sia il più piccolo possibile. A tal fine, calcoliamo il gradiente di . Usando la simmetria di abbiamo
Il gradiente punta nella direzione di ascesa più ripida, il che significa che il suo opposto ci dà la direzione in cui la funzione decresce di più: la direzione della discesa più ripida. Alla nostra stima iniziale , dove , abbiamo che Quindi la funzione decresce di più nella direzione del residuo Pertanto, la nostra scelta iniziale beneficerebbe maggiormente dell'aggiunta del vettore per qualche scalare .
Nel passo successivo, scegliamo di nuovo un vettore e aggiungiamo il suo valore all'approssimazione corrente. Usando lo stesso argomento di prima scegliamo per qualche scalare . Continuiamo in questo modo, cosicché la -esima iterazione del nostro vettore è
In modo equivalente, vogliamo costruire lo spazio da cui scegliamo le nostre stime migliorate aggiungendo , , e così via, in ordine. Il -esimo vettore stimato appartiene a
Ora, usando la relazione che
vediamo che
Cioè, lo spazio che costruiamo che approssima più efficientemente la soluzione corretta è esattamente lo spazio costruito dall'applicazione successiva della matrice su Un sottospazio di Krylov è lo spazio generato dai vettori delle direzioni successive di discesa più ripida.
Infine ripetiamo che non abbiamo fatto alcuna affermazione numerica sulla scalabilità di questo approccio, né abbiamo discusso il beneficio comparativo per le matrici sparse. Questo è solo inteso a motivare l'uso dei metodi per sottospazi di Krylov e ad aggiungere qualche senso intuitivo per essi. Esploreremo ora il comportamento di questi metodi numericamente.
Verifica della comprensione
Nel flusso di lavoro sopra, abbiamo proposto di minimizzare la norma- dell'errore. Quali altre quantità si potrebbe considerare di minimizzare nella ricerca dello stato fondamentale e del suo autovalore?
Answer
Si potrebbe immaginare di usare il vettore residuo al posto della norma- dell'errore. Potrebbero esserci casi in cui considerare lo stesso vettore di errore è utile.
2. Metodi di Krylov nel calcolo classico
In questa sezione implementiamo le iterazioni di Arnoldi computazionalmente per sfruttare un sottospazio di Krylov nella risoluzione di problemi agli autovalori. Applicheremo prima questo a un esempio su piccola scala, poi esamineremo come il tempo di calcolo scala al crescere della dimensione della matrice di interesse. Un'idea chiave qui è che la generazione dei vettori che spaziano lo spazio di Krylov sarà un grande contributo al tempo di calcolo totale richiesto. La memoria necessaria varia tra i diversi metodi di Krylov. Ma i limiti di memoria possono limitare l'uso dei metodi di Krylov tradizionali.
2.1 Semplice esempio su piccola scala
Nel processo di creazione di un sottospazio di Krylov, dovremo ortonormalizzare i vettori nel nostro sottospazio. Definiamo una funzione che prende un vettore già stabilito nel nostro sottospazio vknown (non si assume normalizzato) e un vettore candidato da aggiungere al nostro sottospazio vnext, e rende vnext ortogonale a vknown e normalizzato. Definiamo inoltre una funzione che itera questo processo per tutti i vettori già stabiliti nel nostro sottospazio di Krylov per garantire un insieme completamente ortonormale.
# vknown is some established vector in our subspace. vnext is one we wish to add,
# which must be orthogonal to vknown.
def orthog_pair(vknown, vnext):
vknown = vknown / np.sqrt(vknown.T @ vknown)
diffvec = vknown.T @ vnext * vknown
vnext = vnext - diffvec
return vnext
# v is the candidate vector to be added to our subspace. s is the existing subspace.
def orthoset(v, s):
v = v / np.sqrt(v.T @ v)
temp = v
for i in range(len(s)):
temp = orthog_pair(s[i], temp)
v = temp / np.sqrt(temp.T @ temp)
return v
Definiamo ora una funzione che costruisce un sottospazio di Krylov iterativamente sempre più grande, finché lo spazio dei vettori di Krylov non copre l'intero spazio della matrice originale. Questo ci permetterà di vedere quanto bene gli autovalori ottenuti con il nostro metodo del sottospazio di Krylov corrispondono ai valori esatti, in funzione della dimensione del sottospazio di Krylov. È importante notare che la nostra funzione krylov_full_build restituisce i vettori di Krylov, gli Hamiltoniani proiettati, gli autovalori e il tempo richiesto.
# Necessary imports and definitions to track time in microseconds
import time
def time_mus():
return int(time.time() * 1000000)
# This function constructs a Krylov subspace that spans the whole space of the original matrix.
# Input:
# v0 : initial vector
# matrix : original matrix to be diagonalized
# Output:
# ks : Krylov vectors
# Hs : projected Hamiltonians
# eigs : eigenvalues
# k_tot_times : time required for the operation
def krylov_full_build(v0, matrix):
t0 = time_mus()
b = v0 / np.sqrt(v0 @ v0.T)
A = matrix
ks = []
ks.append(b)
Hs = []
eigs = []
Hs.append(b.T @ A @ b)
eigs.append(np.array([b.T @ A @ b]))
k_tot_times = []
for j in range(len(A) - 1):
vec = A @ ks[j].T
ortho = orthoset(vec, ks)
ks.append(ortho)
ksarray = np.array(ks)
Hs.append(ksarray @ A @ ksarray.T)
eigs.append(np.linalg.eig(Hs[j + 1]).eigenvalues)
k_tot_times.append(time_mus() - t0)
# Return the Krylov vectors, the projected Hamiltonians, the eigenvalues,
# and the total time required.
return (ks, Hs, eigs, k_tot_times)
Testeremo questo su una matrice ancora abbastanza piccola, ma più grande di quanto vorremmo fare a mano.
# Define our small test matrix
test_matrix = np.array(
[
[4, -1, 0, 1, 0],
[-1, 4, -1, 2, 1],
[0, -1, 4, 3, 3],
[1, 2, 3, 4, 0],
[0, 1, 3, 0, 4],
]
)
# Give the test matrix and an initial guess as arguments in the function defined above.
# Calculate outputs.
test_ks, test_Hs, test_eigs, text_k_tot_times = krylov_full_build(
np.array([0.5, 0.5, 0, 0.5, 0.5]), test_matrix
)
Possiamo verificare le nostre funzioni assicurandoci che nell'ultimo passo (quando lo spazio di Krylov è l'intero spazio vettoriale della matrice originale) gli autovalori del metodo di Krylov corrispondano esattamente a quelli della diagonalizzazione numerica esatta:
print(np.linalg.eig(test_matrix).eigenvalues)
print(test_eigs[len(test_matrix) - 1])
[-1.36956923 8.43756009 2.9040308 5.34436028 4.68361806]
[-1.36956923 8.43756009 2.9040308 4.68361806 5.34436028]
È stato un successo. Naturalmente, ciò che conta davvero è quanto buona sia la nostra approssimazione in funzione della dimensione del nostro sottospazio di Krylov. Poiché siamo spesso interessati a trovare stati fondamentali e altri autovalori minimi (e per altre ragioni più algebriche spiegate di seguito), esaminiamo la nostra stima dell'autovalore più basso in funzione della dimensione del sottospazio di Krylov. Ovvero
def errors(matrix, krylov_eigs):
targ_min = min(np.linalg.eig(matrix).eigenvalues)
err = []
for i in range(len(matrix)):
err.append(min(krylov_eigs[i]) - targ_min)
return err
import matplotlib.pyplot as plt
krylov_error = errors(test_matrix, test_eigs)
plt.plot(krylov_error)
plt.axhline(y=0, color="red", linestyle="--") # Add dashed red line at y=0
plt.xlabel("Order of Krylov subspace") # Add x-axis label
plt.ylabel("Error in minimum eigenvalue") # Add y-axis label
plt.show()
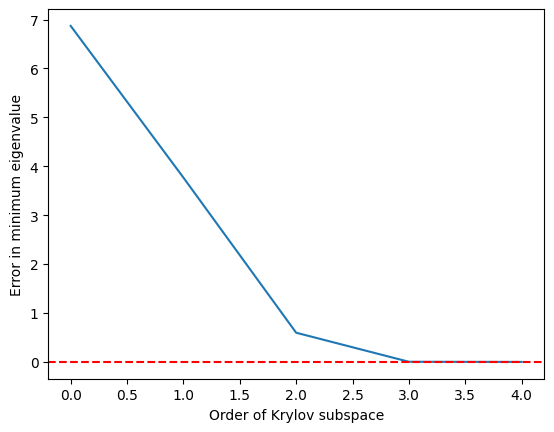
Vediamo che l'autovalore minimo viene raggiunto con buona precisione una volta che il sottospazio di Krylov è cresciuto fino a ed è perfetto a partire da
2.2 Scala temporale con la dimensione della matrice
Convinciamoci che il metodo di Krylov può essere vantaggioso rispetto agli eigensolvers numerici esatti nel modo seguente:
- Costruire matrici casuali (non sparse, non l'applicazione ideale per KQD)
- Determinare gli autovalori usando due metodi: direttamente con NumPy e usando un sottospazio di Krylov.
- Scegliamo un limite di precisione che gli autovalori devono soddisfare prima di accettare le stime di Krylov.
- Confrontare il tempo reale necessario per risolvere nei due modi.
Avvertenze: Come discuteremo in dettaglio di seguito, la diagonalizzazione quantistica di Krylov è meglio applicata a operatori le cui rappresentazioni matriciali sono sparse e/o possono essere scritte usando un piccolo numero di gruppi di operatori di Pauli che commutano. Le matrici casuali che utilizziamo qui non soddisfano questa descrizione. Sono utili solo per sondare la scala a cui i metodi di Krylov classici potrebbero essere vantaggiosi. In secondo luogo, usando il metodo di Krylov calcoleremo gli autovalori usando molti sottospazi di Krylov di dimensioni diverse. Riporteremo il tempo necessario per il sottospazio di Krylov di dimensione minima che raggiunge la precisione richiesta per l'autovalore dello stato fondamentale. Anche questo è un po' diverso dal risolvere un problema intrattabile per gli eigensolvers esatti, poiché usiamo la soluzione esatta per valutare la dimensione necessaria.
Iniziamo generando il nostro insieme di matrici casuali.
import numpy as np
# Set the random seed
np.random.seed(42)
# how many random matrices will we make
num_matrix = 200
matrices = []
for m in range(1, num_matrix):
matrices.append(np.random.rand(m, m))
Ora diagonalizziamo ciascuna matrice direttamente, usando numpy. Calcoliamo il tempo necessario per la diagonalizzazione per un confronto successivo.
matrix_numpy_times = []
matrix_numpy_eigs = []
for mm in range(num_matrix - 1):
t0 = time_mus()
matrix_numpy_eigs.append(min(np.linalg.eig(matrices[mm]).eigenvalues))
matrix_numpy_times.append(time_mus() - t0)
plt.plot(matrix_numpy_times)
plt.xlabel("Dimension of matrix") # Add x-axis label
plt.ylabel("Time to diagonalize (microsec)") # Add y-axis label
plt.show()
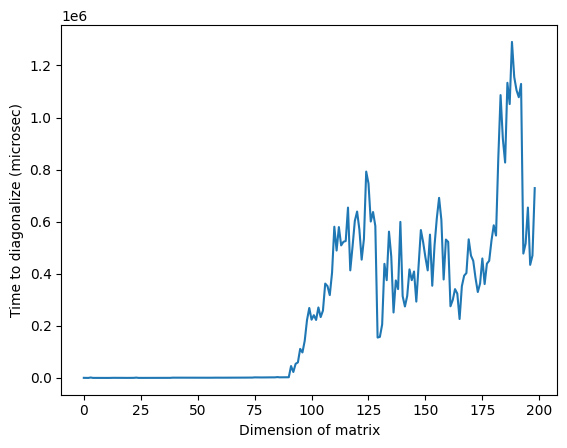
Nota che nell'immagine sopra, il tempo anomalamente alto intorno a una dimensione di 125 potrebbe essere dovuto alla natura casuale delle matrici o all'implementazione sul processore classico utilizzato, ma non è riproducibile. Rieseguendo il codice si otterrà un profilo diverso con picchi anomali diversi.
Ora per ogni matrice costruiremo un sottospazio di Krylov e calcoleremo gli autovalori a passi. Ad ogni passo, verificheremo se l'autovalore più basso è stato ottenuto entro il nostro errore assoluto specificato. Il sottospazio che per primo ci fornisce autovalori entro il nostro errore specificato è quello per cui registreremo i tempi di calcolo. L'esecuzione di questa cella potrebbe richiedere diversi minuti, a seconda della velocità del processore. Sei libero di saltare la valutazione o ridurre la dimensione massima delle matrici diagonalizzate. Guardare i risultati precalcolati è sufficiente.
# Choose the absolute error you can tolerate, and make a list for tracking the Krylov subspace size
# at which that error is achieved.
abserr = 0.05
accept_subspace_size = []
# Lists to store total time spent on the Krylov method, and the subset of that time spent on
# diagonalizing the projected matrix.
matrix_krylov_tot_times = []
matrix_krylov_dim = []
# Step through all our random matrices
for mm in range(0, num_matrix - 1):
test_ks, test_Hs, test_eigs, test_k_tot_times = krylov_full_build(
np.ones(len(matrices[mm])), matrices[mm]
)
# We have not yet found a Krylov subspace that produces our minimum eigenvalue to
# within the required error.
found = 0
for j in range(0, len(matrices[mm]) - 1):
# If we still haven't found the desired subspace...
if found == 0:
# ...but if this one satisfies the requirement, then record everything
if (
abs((min(test_eigs[j]) - matrix_numpy_eigs[mm]) / matrix_numpy_eigs[mm])
< abserr
):
accept_subspace_size.append(j)
matrix_krylov_tot_times.append(test_k_tot_times[j])
matrix_krylov_dim.append(mm)
found = 1
Tracciamo i tempi ottenuti con questi due metodi per un confronto:
plt.plot(matrix_numpy_times, color="blue")
plt.plot(matrix_krylov_dim, matrix_krylov_tot_times, color="green")
plt.xlabel("Dimension of matrix") # Add x-axis label
plt.ylabel("Time to diagonalize (microsec)") # Add y-axis label
plt.show()

Questi sono i tempi reali richiesti, ma ai fini della discussione, levighiamo queste curve mediando su alcuni punti adiacenti / dimensioni di matrice. Questo viene fatto di seguito:
smooth_numpy_times = []
smooth_krylov_times = []
# Choose the number of adjacent points over which to average forward;
# the same will be used backward.
smooth_steps = 10
# We will do this smoothing for all points/matrix dimensions
for i in range(len(matrix_krylov_tot_times)):
# Ensure we don't exceed the boundaries of our lists
start = max(0, i - smooth_steps)
end = min(len(matrix_krylov_tot_times) - 1, i + smooth_steps)
# Dummy variables for accumulating an average over adjacent points. This is done for both Krylov
# and the NumPy calculations.
smooth_count = 0
smooth_numpy_sum = 0
smooth_krylov_sum = 0
for j in range(start, end):
smooth_numpy_sum = smooth_numpy_sum + matrix_numpy_times[j]
smooth_krylov_sum = smooth_krylov_sum + matrix_krylov_tot_times[j]
smooth_count = smooth_count + 1
# Appending the averaged adjacent values to our new smooth lists
smooth_numpy_times.append(smooth_numpy_sum / smooth_count)
smooth_krylov_times.append(smooth_krylov_sum / smooth_count)
plt.plot(smooth_numpy_times, color="blue")
plt.plot(smooth_krylov_times, color="green")
plt.xlabel("Dimension of matrix") # Add x-axis label
plt.ylabel("Time to diagonalize (smoothed, microsec)") # Add y-axis label
plt.show()

Nota che il tempo necessario per la costruzione di un sottospazio di Krylov supera inizialmente il tempo necessario per la diagonalizzazione completa di numpy. Ma man mano che la dimensione della matrice cresce, il metodo di Krylov diventa vantaggioso. Questo vale anche abbassando il nostro errore accettabile, ma il vantaggio si manifesta a una dimensione di matrice maggiore. Vale la pena analizzare questo aspetto.
La complessità temporale della diagonalizzazione numerica è (con alcune variazioni tra gli algoritmi). La complessità temporale della generazione di una base ortonormale di vettori è anch'essa . Quindi il vantaggio del metodo di Krylov non è legato all'uso di base ortonormale, ma all'uso di una particolare base ortonormale che seleziona efficacemente gli autovalori di interesse. Lo abbiamo già visto dalla bozza di dimostrazione nella prima sezione di questa lezione, ed è fondamentale per le garanzie di convergenza nei metodi di Krylov. Ricapitoliamo i progressi finora:
- Per matrici molto grandi, il metodo del sottospazio di Krylov può fornire autovalori approssimati entro le tolleranze richieste più velocemente degli algoritmi di diagonalizzazione tradizionali.
- Per tali matrici molto grandi, la generazione di un sottospazio di Krylov è la parte più dispendiosa in termini di tempo del metodo del sottospazio di Krylov.
- Pertanto, un modo efficiente per generare un sottospazio di Krylov sarebbe di grande valore. È qui che finalmente entra in gioco il computer quantistico.
Verifica la tua comprensione
Fai riferimento al grafico levigato dei tempi di diagonalizzazione rispetto alla dimensione della matrice sopra.
(a) A quale dimensione di matrice circa il metodo di Krylov è diventato più veloce, secondo questo grafico?
(b) Quali aspetti del calcolo potrebbero cambiare la dimensione a cui il metodo di Krylov diventa più veloce?
Answer
(a) Le risposte possono variare se riesegui il calcolo, ma il metodo di Krylov diventa più veloce a circa dimensione 80-85.
(b) Ci sono molte possibili risposte. Alcuni fattori importanti sono la precisione che richediamo e la sparsità delle matrici da diagonalizzare.
3. Krylov tramite evoluzione temporale
Tutto ciò che abbiamo descritto finora può essere fatto classicamente. Quindi come e quando useremmo un computer quantistico? Per matrici molto grandi, il metodo di Krylov può richiedere lunghi tempi di calcolo e grandi quantità di memoria. Il tempo necessario per l'operazione matriciale di su scala come nel caso peggiore. Anche la moltiplicazione di matrici sparse su un vettore (il caso tipico per i solutori di Krylov classici) ha una complessità temporale che scala come . Questo viene fatto per ogni vettore che vogliamo nel nostro sottospazio. La dimensione del sottospazio di solito non è una frazione significativa di , e spesso scala come . Quindi la generazione di tutti i vettori scala come nel caso peggiore. Sebbene ci siano altri passaggi, come l'ortogonalizzazione, questa è la scala dominante da tenere a mente.
Il calcolo quantistico ci permette di cambiare quali attributi del problema determinano la scala del tempo e delle risorse necessarie. Invece della dipendenza dalla dimensione della matrice in generale, vedremo cose come il numero di shots e il numero di termini di Pauli non commutanti che compongono l'Hamiltoniano. Vediamo come funziona.
3.1 Evoluzione temporale
Ricorda che l'operatore che fa evolvere temporalmente uno stato quantistico è (ed è molto comune, specialmente nel calcolo quantistico, omettere dalla notazione). Un modo di comprendere e persino realizzare tale funzione esponenziale di un operatore è guardare la sua espansione in serie di Taylor. Nota che questa operazione applicata a qualche vettore iniziale produce una somma di termini con potenze crescenti di applicate allo stato iniziale. Sembra che possiamo costruire il nostro sottospazio di Krylov semplicemente facendo evolvere temporalmente il nostro stato iniziale!
Il problema sta nel realizzare l'evoluzione temporale su un vero computer quantistico. Molti dei termini nell'Hamiltoniano non commutano tra loro. Quindi, mentre alcuni semplici operatori esponenziali come corrispondono a circuiti semplici, gli Hamiltoniani generali non lo fanno. E poiché contengono termini non commutanti, non possiamo semplicemente decomporre l'esponenziale in un prodotto di quelli semplici, come si può fare con i numeri.
Quindi questo non è banale, ma è un processo ben studiato nel calcolo quantistico. Eseguiamo l'evoluzione temporale sui computer quantistici usando un processo chiamato trotterizzazione, che di per sé è un argomento ricco[10]. Ma a un livello molto alto, suddividendo l'evoluzione temporale in passi molto piccoli, diciamo passi di dimensione , limitiamo gli effetti della non-commutatività dei termini.
dove .
Chiamiamo un sottospazio di Krylov di ordine r che abbiamo generato nel contesto classico usando direttamente le potenze di H un "sottospazio di Krylov potenza".
Ora generiamo uno spazio simile usando l'operatore unitario di evoluzione temporale ; lo chiameremo "spazio di Krylov unitario" . Il sottospazio di Krylov potenza che usiamo classicamente non può essere generato direttamente su un computer quantistico poiché non è un operatore unitario. L'uso del sottospazio di Krylov unitario può essere dimostrato fornire garanzie di convergenza simili a quelle del sottospazio di Krylov potenza, ovvero, l'errore dello stato fondamentale converge in modo efficiente purché lo stato iniziale abbia una sovrapposizione con il vero stato fondamentale che non sia esponenzialmente vanescente, e purché vi sia un gap sufficiente tra gli autovalori. Vedi Ref [1] per una discussione più precisa della convergenza.
Qui, le potenze di diventano diversi passi temporali (la potenza di corrisponde ad avanzare di un tempo ). Possiamo etichettare l'elemento del sottospazio che è evoluto temporalmente per un tempo totale come .
Possiamo proiettare il nostro Hamiltoniano H sul sottospazio di Krylov unitario, . In altre parole, calcoliamo ciascun elemento della matrice di nella base . Chiameremo questa matrice proiettata .
3.2 Come implementare su un computer quantistico
Gli elementi della matrice di sono dati dai valori di aspettazione , che possono essere stimati usando il computer quantistico. Tieni presente che può essere scritto come una somma di operatori di Pauli su diversi qubit, e che non tutti gli operatori di Pauli possono essere misurati simultaneamente. Possiamo ordinare i termini di Pauli in gruppi di termini commutanti e misurarli tutti insieme. Ma potremmo aver bisogno di molti tali gruppi per coprire tutti i termini. Quindi il numero di gruppi commutanti distinti in cui i termini possono essere partizionati, , diventa importante.
Qui è una stringa di Pauli della forma o un insieme di tali stringhe di Pauli che commutano tra loro. Dato che possiamo scrivere come tale somma di operatori misurabili, le seguenti espressioni per gli elementi della matrice di possono essere realizzate usando la primitiva Estimator di Qiskit Runtime.
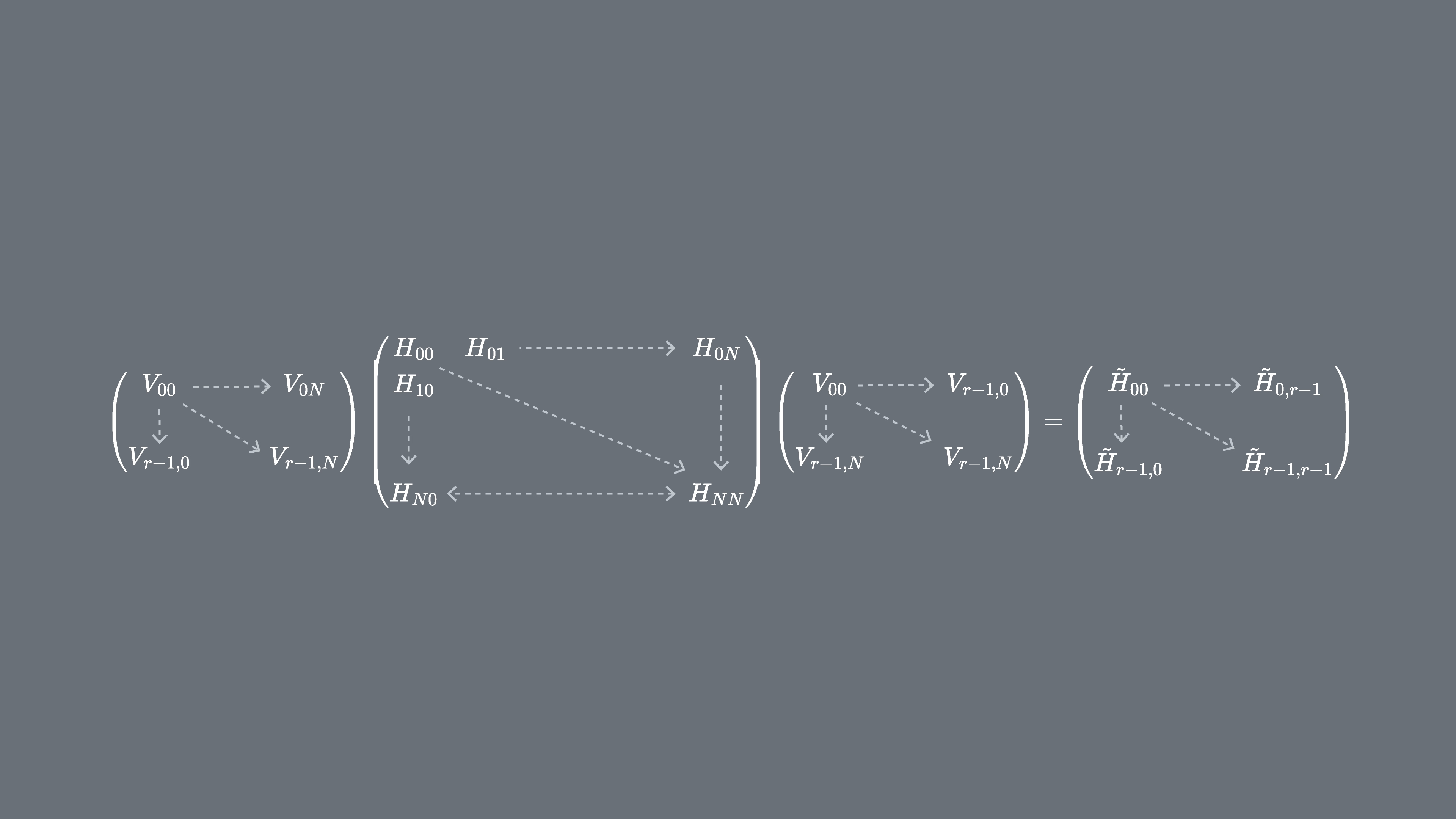
Dove sono i vettori dello spazio di Krylov unitario e sono i multipli del passo temporale scelto. Su un computer quantistico, il calcolo di ciascun elemento della matrice può essere eseguito con qualsiasi algoritmo che ci permette di ottenere la sovrapposizione tra stati quantistici. In questa lezione ci concentreremo sul test di Hadamard. Dato che ha dimensione , l'Hamiltoniano proiettato nel sottospazio avrà dimensioni . Con sufficientemente piccolo (generalmente è sufficiente per ottenere la convergenza delle stime degli autovalori) possiamo quindi diagonalizzare facilmente l'Hamiltoniano proiettato classicamente. Tuttavia, non possiamo diagonalizzare direttamente a causa della non-ortogonalità dei vettori dello spazio di Krylov. Dovremo misurare le loro sovrapposizioni e costruire una matrice
Questo ci permette di risolvere il problema agli autovalori in uno spazio non ortogonale (chiamato anche problema generalizzato agli autovalori)
Si possono quindi ottenere stime degli autovalori e degli autostati di esaminando le soluzioni di questo problema generalizzato agli autovalori. Ad esempio, la stima dell'energia dello stato fondamentale si ottiene prendendo il più piccolo autovalore e lo stato fondamentale dal corrispondente autovettore . I coefficienti in determinano il contributo dei diversi vettori che spaziano .
Problema generalizzato agli autovalori
Perché non possiamo semplicemente diagonalizzare ? Poiché contiene le informazioni sulla geometria della base di Krylov (che è non-ortogonale in tutti i casi eccetto quelli molto speciali), da solo non descrive una proiezione dell'Hamiltoniano completo, quindi i suoi autovalori non hanno una relazione particolare con quelli dell'Hamiltoniano completo — potrebbero essere qualsiasi valore casuale. Risolvere il problema generalizzato agli autovalori è necessario per ottenere gli autovalori e gli autovettori approssimati corrispondenti alla proiezione dell'Hamiltoniano completo nello spazio di Krylov.
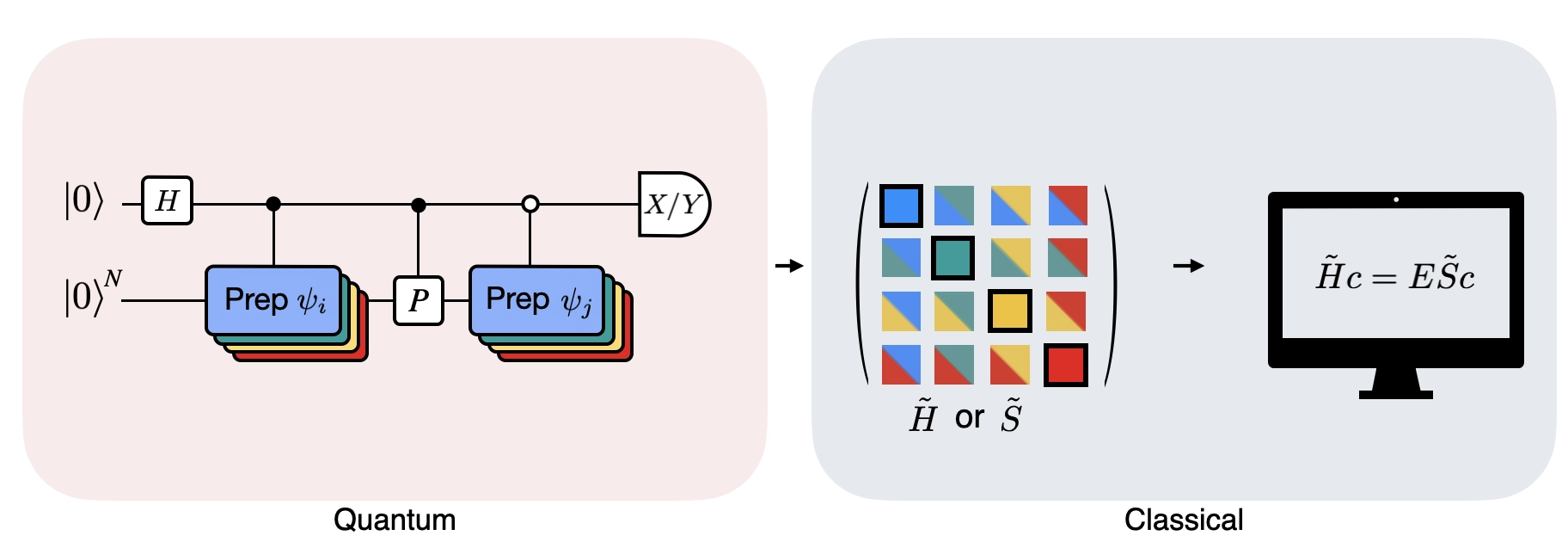
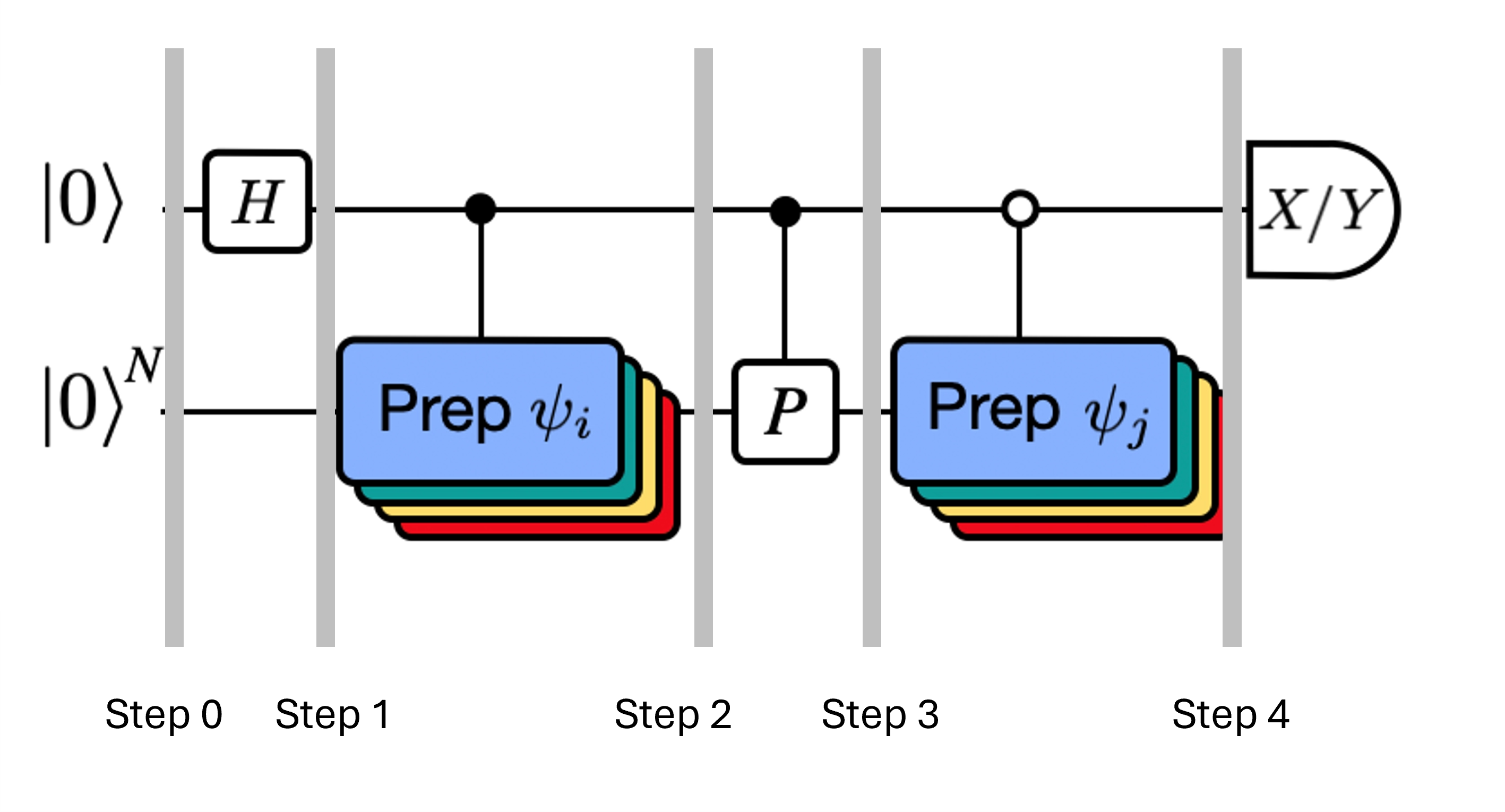
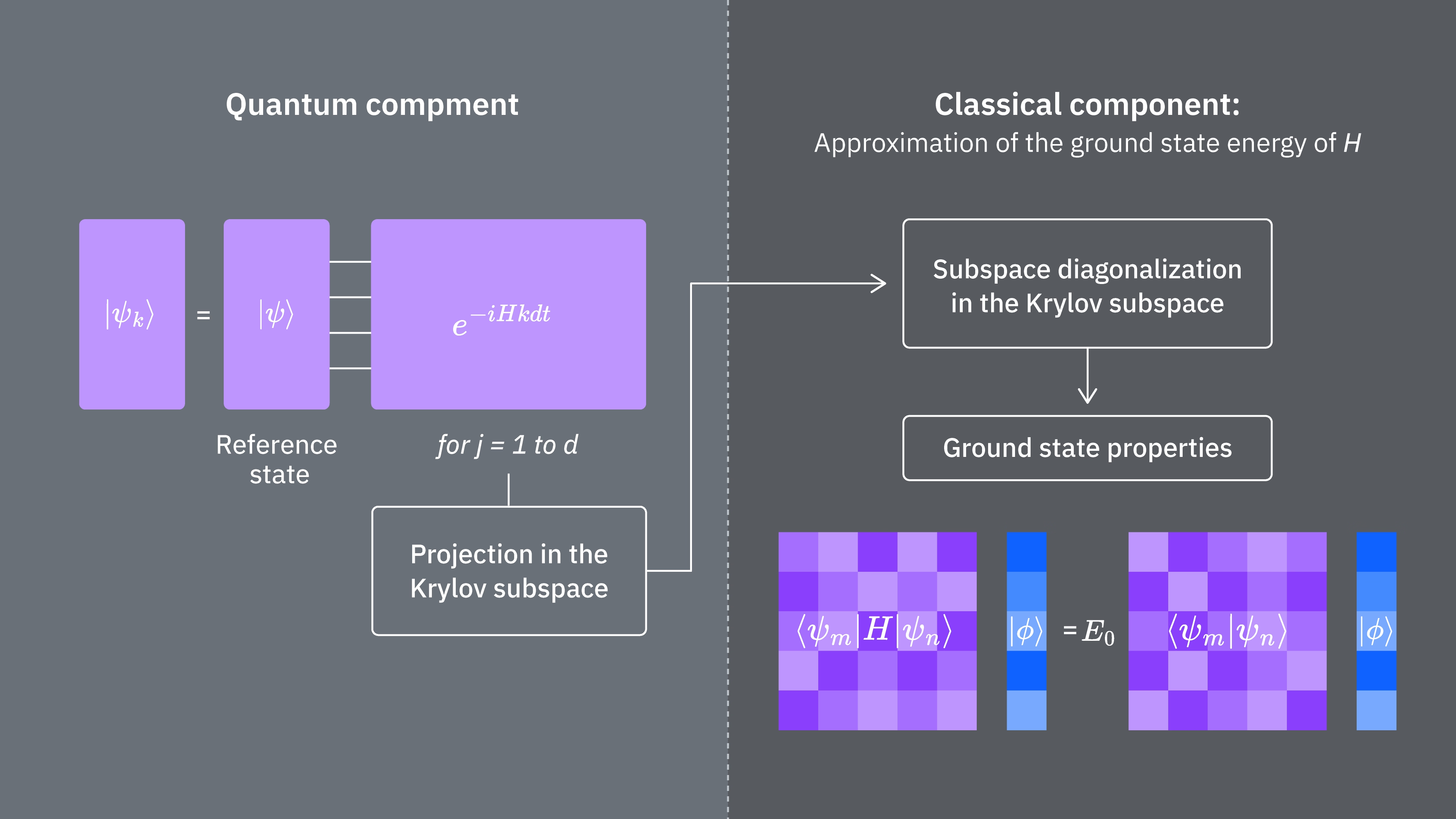
La figura mostra una rappresentazione circuitale del test di Hadamard modificato, un metodo usato per calcolare la sovrapposizione tra diversi stati quantistici. Per ciascun elemento della matrice , viene eseguito un test di Hadamard tra lo stato , . Questo è evidenziato nella figura dallo schema cromatico per gli elementi della matrice e le corrispondenti operazioni , . Pertanto, è necessario un insieme di test di Hadamard per tutte le possibili combinazioni di vettori dello spazio di Krylov per calcolare tutti gli elementi della matrice dell'Hamiltoniano proiettato . Il filo superiore nel circuito del test di Hadamard è un qubit ancilla che viene misurato nella base X o Y; il suo valore di aspettazione determina il valore della sovrapposizione tra gli stati. Il filo inferiore rappresenta tutti i qubit dell'Hamiltoniano del sistema. L'operazione prepara il qubit del sistema nello stato controllato dallo stato del qubit ancilla (analogamente per ) e l'operazione rappresenta la decomposizione di Pauli dell'Hamiltoniano del sistema . L'implementazione di questo su un computer quantistico è mostrata in maggior dettaglio di seguito.
4. Diagonalizzazione quantistica di Krylov su un computer quantistico
Implementeremo ora la diagonalizzazione quantistica di Krylov su un vero computer quantistico. Iniziamo importando alcuni pacchetti utili.
import numpy as np
import scipy as sp
import matplotlib.pylab as plt
from typing import Union, List
import warnings
from qiskit.quantum_info import SparsePauliOp, Pauli
from qiskit.circuit import Parameter
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.circuit.library import PauliEvolutionGate
from qiskit.synthesis import LieTrotter
# from qiskit.providers.fake_provider import Fake20QV1
from qiskit_ibm_runtime import QiskitRuntimeService, EstimatorV2 as Estimator, Batch
import itertools as it
warnings.filterwarnings("ignore")
Definiamo la funzione seguente per risolvere il problema generalizzato degli autovalori appena illustrato sopra.
def solve_regularized_gen_eig(
h: np.ndarray,
s: np.ndarray,
threshold: float,
k: int = 1,
return_dimn: bool = False,
) -> Union[float, List[float]]:
"""
Method for solving the generalized eigenvalue problem with regularization
Args:
h (numpy.ndarray):
The effective representation of the matrix in our Krylov subspace
s (numpy.ndarray):
The matrix of overlaps between vectors of our Krylov subspace
threshold (float):
Cut-off value for the eigenvalue of s
k (int):
Number of eigenvalues to return
return_dimn (bool):
Whether to return the size of the regularized subspace
Returns:
lowest k-eigenvalue(s) that are the solution of the regularized generalized eigenvalue problem
"""
s_vals, s_vecs = sp.linalg.eigh(s)
s_vecs = s_vecs.T
good_vecs = np.array([vec for val, vec in zip(s_vals, s_vecs) if val > threshold])
h_reg = good_vecs.conj() @ h @ good_vecs.T
s_reg = good_vecs.conj() @ s @ good_vecs.T
if k == 1:
if return_dimn:
return sp.linalg.eigh(h_reg, s_reg)[0][0], len(good_vecs)
else:
return sp.linalg.eigh(h_reg, s_reg)[0][0]
else:
if return_dimn:
return sp.linalg.eigh(h_reg, s_reg)[0][:k], len(good_vecs)
else:
return sp.linalg.eigh(h_reg, s_reg)[0][:k]
Almeno nella fase iniziale di benchmarking, è utile conoscere una soluzione classica esatta per verificare il comportamento di convergenza. La funzione seguente calcola l'energia dello stato fondamentale di un'Hamiltoniana, usando l'Hamiltoniana e il numero di qubit come argomenti.
def single_particle_gs(H_op, n_qubits):
"""
Find the ground state of the single particle(excitation) sector
"""
H_x = []
for p, coeff in H_op.to_list():
H_x.append(set([i for i, v in enumerate(Pauli(p).x) if v]))
H_z = []
for p, coeff in H_op.to_list():
H_z.append(set([i for i, v in enumerate(Pauli(p).z) if v]))
H_c = H_op.coeffs
print("n_sys_qubits", n_qubits)
n_exc = 1
sub_dimn = int(sp.special.comb(n_qubits + 1, n_exc))
print("n_exc", n_exc, ", subspace dimension", sub_dimn)
few_particle_H = np.zeros((sub_dimn, sub_dimn), dtype=complex)
sparse_vecs = [
set(vec) for vec in it.combinations(range(n_qubits + 1), r=n_exc)
] # list all of the possible sets of n_exc indices of 1s in n_exc-particle states
m = 0
for i, i_set in enumerate(sparse_vecs):
for j, j_set in enumerate(sparse_vecs):
m += 1
if len(i_set.symmetric_difference(j_set)) <= 2:
for p_x, p_z, coeff in zip(H_x, H_z, H_c):
if i_set.symmetric_difference(j_set) == p_x:
sgn = ((-1j) ** len(p_x.intersection(p_z))) * (
(-1) ** len(i_set.intersection(p_z))
)
else:
sgn = 0
few_particle_H[i, j] += sgn * coeff
gs_en = min(np.linalg.eigvalsh(few_particle_H))
print("single particle ground state energy: ", gs_en)
return gs_en
4.1 Passo 1: Mappare il problema su circuiti e operatori quantistici
Definiamo ora un'Hamiltoniana. Questa è distinta dalla funzione precedente nel senso che quella funzione prende un'Hamiltoniana come argomento e restituisce solo lo stato fondamentale, e lo fa in modo classico. L'Hamiltoniana che definiamo qui determina i livelli energetici di tutti gli autostati di energia, e può essere costruita usando operatori di Pauli e implementata su un computer quantistico.
Scegliamo un'Hamiltoniana corrispondente a una catena di spin che possono avere qualsiasi orientazione nello spazio, chiamata "catena di Heisenberg". Assumiamo che l' spin possa essere influenzato dai suoi vicini più prossimi (gli spin e ) ma non dai vicini più lontani. Ammettiamo anche la possibilità che l'interazione tra spin sia diversa quando gli spin puntano lungo assi diversi. Questa asimmetria emerge talvolta, ad esempio, a causa della struttura del reticolo cristallino in cui gli spin sono immersi.
# Define problem Hamiltonian.
n_qubits = 10
# coupling strength for XX, YY, and ZZ interactions
JX = 1
JY = 3
JZ = 2
# Define the Hamiltonian:
H_int = [["I"] * n_qubits for _ in range(3 * (n_qubits - 1))]
for i in range(n_qubits - 1):
H_int[i][i] = "Z"
H_int[i][i + 1] = "Z"
for i in range(n_qubits - 1):
H_int[n_qubits - 1 + i][i] = "X"
H_int[n_qubits - 1 + i][i + 1] = "X"
for i in range(n_qubits - 1):
H_int[2 * (n_qubits - 1) + i][i] = "Y"
H_int[2 * (n_qubits - 1) + i][i + 1] = "Y"
H_int = ["".join(term) for term in H_int]
H_tot = [
(term, JZ)
if term.count("Z") == 2
else (term, JY)
if term.count("Y") == 2
else (term, JX)
for term in H_int
]
# Get operator
H_op = SparsePauliOp.from_list(H_tot)
print(H_tot)
[('ZZIIIIIIII', 2), ('IZZIIIIIII', 2), ('IIZZIIIIII', 2), ('IIIZZIIIII', 2), ('IIIIZZIIII', 2), ('IIIIIZZIII', 2), ('IIIIIIZZII', 2), ('IIIIIIIZZI', 2), ('IIIIIIIIZZ', 2), ('XXIIIIIIII', 1), ('IXXIIIIIII', 1), ('IIXXIIIIII', 1), ('IIIXXIIIII', 1), ('IIIIXXIIII', 1), ('IIIIIXXIII', 1), ('IIIIIIXXII', 1), ('IIIIIIIXXI', 1), ('IIIIIIIIXX', 1), ('YYIIIIIIII', 3), ('IYYIIIIIII', 3), ('IIYYIIIIII', 3), ('IIIYYIIIII', 3), ('IIIIYYIIII', 3), ('IIIIIYYIII', 3), ('IIIIIIYYII', 3), ('IIIIIIIYYI', 3), ('IIIIIIIIYY', 3)]
Il codice seguente limita l'Hamiltoniana agli stati a singola particella e usa la norma spettrale per impostare una buona dimensione per il passo temporale . Scegliamo euristicamente un valore per il passo temporale dt (basato su limiti superiori della norma dell'Hamiltoniana). Il Rif. [9] ha dimostrato che un passo temporale sufficientemente piccolo è , e che è preferibile, entro certi limiti, sottostimare questo valore piuttosto che sovrastimarlo, poiché una sovrastima può permettere contributi degli stati ad alta energia di corrompere persino lo stato ottimale nello spazio di Krylov. D'altra parte, scegliere troppo piccolo porta a un peggior condizionamento del sottospazio di Krylov, poiché i vettori della base di Krylov differiscono meno da un passo temporale all'altro.
# Get Hamiltonian restricted to single-particle states
single_particle_H = np.zeros((n_qubits, n_qubits))
for i in range(n_qubits):
for j in range(i + 1):
for p, coeff in H_op.to_list():
p_x = Pauli(p).x
p_z = Pauli(p).z
if all(p_x[k] == ((i == k) + (j == k)) % 2 for k in range(n_qubits)):
sgn = ((-1j) ** sum(p_z[k] and p_x[k] for k in range(n_qubits))) * (
(-1) ** p_z[i]
)
else:
sgn = 0
single_particle_H[i, j] += sgn * coeff
for i in range(n_qubits):
for j in range(i + 1, n_qubits):
single_particle_H[i, j] = np.conj(single_particle_H[j, i])
# Set dt according to spectral norm
dt = np.pi / np.linalg.norm(single_particle_H, ord=2)
dt
np.float64(0.17453292519943295)
Specifichiamo il numero di passi di Trotter da utilizzare nell'evoluzione temporale. Specifichiamo inoltre una dimensione massima di Krylov pari a 4. Questa dimensione di Krylov non è sufficientemente grande per applicazioni realistiche. Ma è sufficiente per questo esempio. Inoltre, verificheremo la convergenza anche a dimensioni inferiori. Nei prossimi capitoli esploreremo metodi che ci permettono di scalare e proiettare le nostre Hamiltoniane su sottospazi più grandi.
# Set parameters for quantum Krylov algorithm
krylov_dim = 4 # size of krylov subspace
num_trotter_steps = 4
dt_circ = dt / num_trotter_steps
Preparazione dello stato
Scegliamo uno stato di riferimento che abbia una certa sovrapposizione con lo stato fondamentale. Per questa Hamiltoniana, utilizziamo uno stato con un'eccitazione nel qubit centrale come nostro stato di riferimento.
qc_state_prep = QuantumCircuit(n_qubits)
qc_state_prep.x(int(n_qubits / 2) + 1)
qc_state_prep.draw("mpl", scale=0.5)

Evoluzione temporale
Possiamo realizzare l'operatore di evoluzione temporale generato da una data Hamiltoniana: tramite l'approssimazione di Lie-Trotter. Per semplicità utilizziamo il PauliEvolutionGate integrato nel circuito di evoluzione temporale. La sintassi generale è la seguente.
t = Parameter("t")
## Create the time-evo op circuit
evol_gate = PauliEvolutionGate(
H_op, time=t, synthesis=LieTrotter(reps=num_trotter_steps)
)
qr = QuantumRegister(n_qubits)
qc_evol = QuantumCircuit(qr)
qc_evol.append(evol_gate, qargs=qr)
<qiskit.circuit.instructionset.InstructionSet at 0x7ccaa4664250>
Utilizzeremo una versione di questo nel test di Hadamard di seguito, avanzando di passi temporali .
Test di Hadamard
Ricorda che vogliamo calcolare gli elementi di matrice sia di sia della matrice di Gram usando il test di Hadamard. Rivediamo come funziona in questo contesto, concentrandoci prima sulla costruzione di L'intero processo è rappresentato graficamente di seguito. I livelli dei blocchi colorati di preparazione dello stato ricordano che questo processo viene eseguito per tutte le combinazioni di e nel nostro sottospazio.
Gli stati del sistema nei passi indicati sono:
Qui è un termine di Pauli nella decomposizione dell'Hamiltoniana (nota che non può essere una combinazione lineare di più termini di Pauli commutanti poiché non sarebbe unitaria — il raggruppamento è possibile usando una costruzione diversa che mostreremo in seguito). , sono operazioni controllate che preparano i vettori , dello spazio di Krylov unitario, con . Applicando misurazioni di e a questo circuito si calcolano rispettivamente le parti reale e immaginaria degli elementi di matrice richiesti.
Partendo dal Passo 4 sopra, applichiamo il gate di Hadamard al qubit zero.
Poi misuriamo oppure .
Dall'identità . Analogamente, misurare dà
Aggiungendo questi passi all'evoluzione temporale impostata in precedenza, scriviamo quanto segue.
## Create the time-evo op circuit
evol_gate = PauliEvolutionGate(
H_op, time=dt, synthesis=LieTrotter(reps=num_trotter_steps)
)
## Create the time-evo op dagger circuit
evol_gate_d = PauliEvolutionGate(
H_op, time=dt, synthesis=LieTrotter(reps=num_trotter_steps)
)
evol_gate_d = evol_gate_d.inverse()
# Put pieces together
qc_reg = QuantumRegister(n_qubits)
qc_temp = QuantumCircuit(qc_reg)
qc_temp.compose(qc_state_prep, inplace=True)
for _ in range(num_trotter_steps):
qc_temp.append(evol_gate, qargs=qc_reg)
for _ in range(num_trotter_steps):
qc_temp.append(evol_gate_d, qargs=qc_reg)
qc_temp.compose(qc_state_prep.inverse(), inplace=True)
# Create controlled version of the circuit
controlled_U = qc_temp.to_gate().control(1)
# Create hadamard test circuit for real part
qr = QuantumRegister(n_qubits + 1)
qc_real = QuantumCircuit(qr)
qc_real.h(0)
qc_real.append(controlled_U, list(range(n_qubits + 1)))
qc_real.h(0)
print("Circuit for calculating the real part of the overlap in S via Hadamard test")
qc_real.draw("mpl", fold=-1, scale=0.5)
Circuit for calculating the real part of the overlap in S via Hadamard test

Avevamo già avvertito della profondità coinvolta nei circuiti di Trotter. Eseguire il test di Hadamard in queste condizioni può produrre un circuito ancora più profondo, specialmente una volta decomposto in gate nativi. Questo aumenterà ulteriormente se teniamo conto della topologia del dispositivo. Quindi, prima di utilizzare tempo su un computer quantistico, è una buona idea verificare la profondità a 2 qubit del nostro circuito.
print(
"Number of layers of 2Q operations",
qc_real.decompose(reps=2).depth(lambda x: x[0].num_qubits == 2),
)
Number of layers of 2Q operations 14401
Un circuito di questa profondità non può restituire risultati utilizzabili sui computer quantistici moderni. Se vogliamo costruire e abbiamo bisogno di un approccio migliore. Questo è il motivo per cui viene introdotto di seguito il test di Hadamard efficiente.
4. 2 Passo 2. Ottimizzare i circuiti e gli operatori per l'hardware target
Test di Hadamard efficiente
Possiamo ottimizzare i circuiti profondi per il test di Hadamard che abbiamo ottenuto introducendo alcune approssimazioni e facendo affidamento su alcune ipotesi sul Hamiltoniano del modello. Ad esempio, considera il seguente circuito per il test di Hadamard:
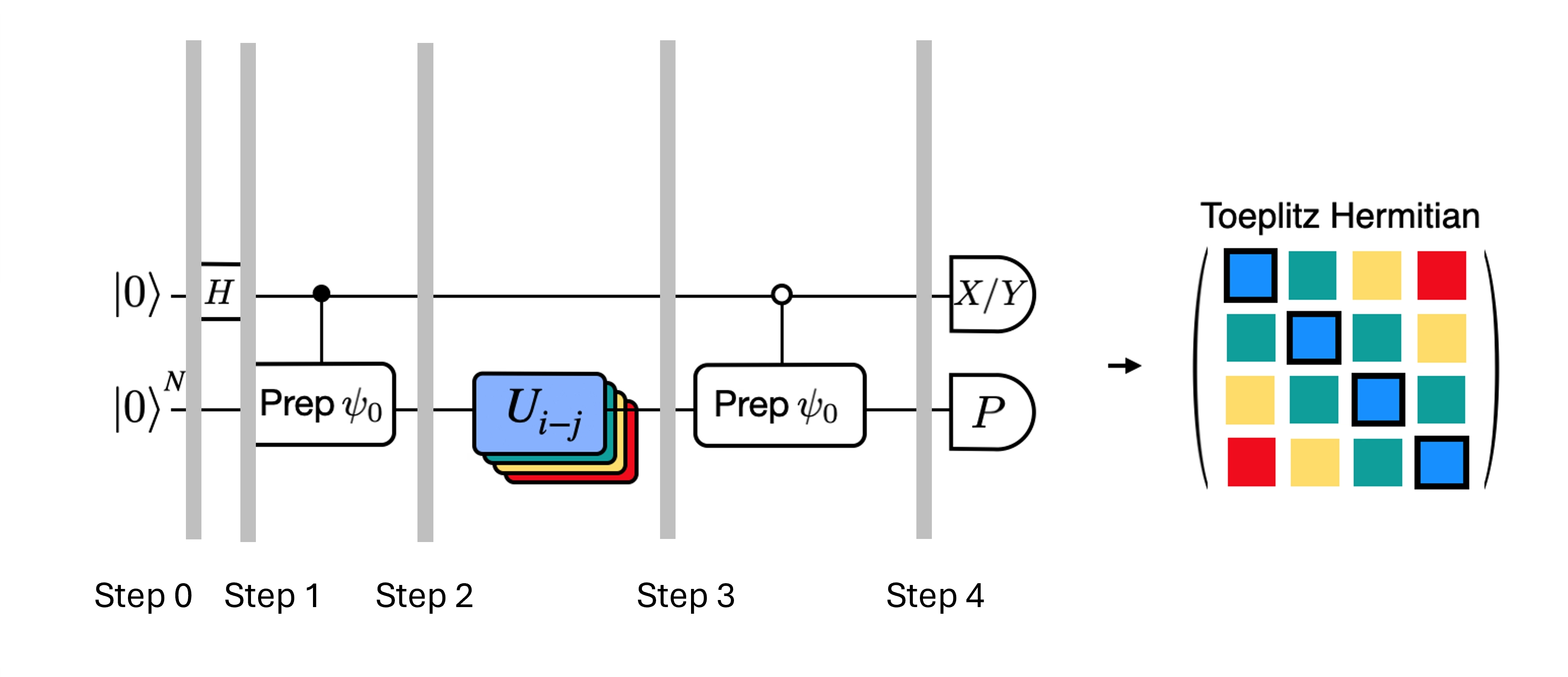
Supponiamo di poter calcolare classicamente , l'autovalore di sotto l'Hamiltoniano . Questa condizione è soddisfatta quando l'Hamiltoniano conserva la simmetria U(1). Sebbene questo possa sembrare un'ipotesi forte, esistono molti casi in cui è lecito assumere la presenza di uno stato vuoto (in questo caso corrispondente allo stato ) che non è influenzato dall'azione dell'Hamiltoniano. Ciò vale ad esempio per gli Hamiltoniani di chimica che descrivono molecole stabili (dove il numero di elettroni è conservato). Dato che il gate prepara lo stato di riferimento desiderato , ad esempio per preparare lo stato HF per la chimica sarebbe un prodotto di NOT a singolo qubit, quindi il controllato è semplicemente un prodotto di CNOT. Il circuito sopra implementa quindi il seguente stato prima della misurazione:
dove abbiamo utilizzato lo sfasamento simulabile classicamente dal passo 2 al 3. Pertanto i valori di aspettazione sono
Grazie a queste ipotesi siamo stati in grado di esprimere i valori di aspettazione degli operatori di interesse con un numero ridotto di operazioni controllate. In effetti, dobbiamo implementare solo la preparazione dello stato controllata e non le evoluzioni temporali controllate. Riformulare il calcolo come sopra ci permetterà di ridurre notevolmente la profondità dei circuiti risultanti.
Nota che, come bonus, poiché l'operatore di Pauli appare ora come una misurazione alla fine del circuito anziché come gate controllato nel mezzo, può essere misurato insieme ad altri operatori di Pauli commutanti come nella decomposizione fornita sopra.
Decomposizione dell'operatore di evoluzione temporale con la decomposizione di Trotter
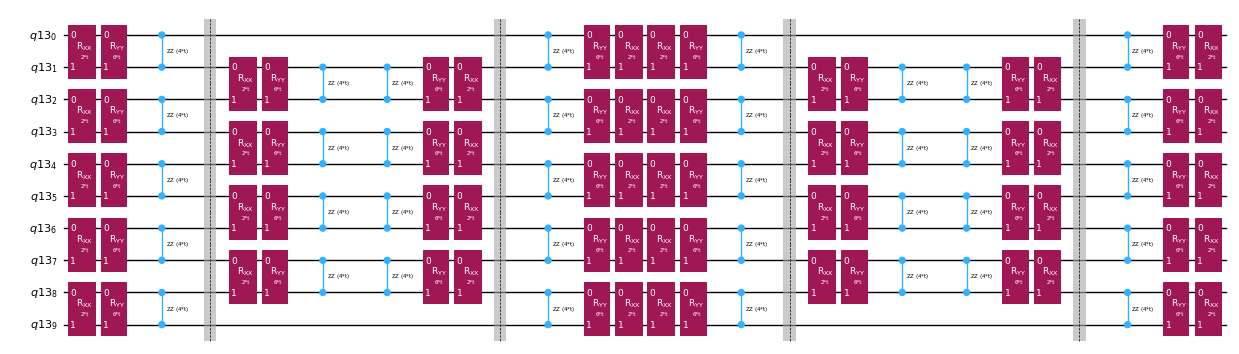
Invece di implementare l'operatore di evoluzione temporale in modo esatto, possiamo usare la decomposizione di Trotter per implementarne un'approssimazione. Ripetere più volte una decomposizione di Trotter di un certo ordine consente di ridurre ulteriormente l'errore introdotto dall'approssimazione. Di seguito, costruiamo direttamente l'implementazione di Trotter nel modo più efficiente per il grafo delle interazioni dell'Hamiltoniano che stiamo considerando (solo interazioni tra vicini più prossimi). In pratica inseriamo rotazioni di Pauli , , con costanti di accoppiamento e e un angolo parametrizzato , che corrispondono all'implementazione approssimata di . Data la differenza di definizione tra le rotazioni di Pauli e l'evoluzione temporale che vogliamo implementare, dovremo usare il parametro per ottenere un'evoluzione temporale di . Inoltre, invertiamo l'ordine delle operazioni per i passi di Trotter con numero dispari di ripetizioni, il che è funzionalmente equivalente ma permette di sintetizzare operazioni adiacenti in un unico unitario . Questo produce un circuito molto meno profondo rispetto a quello ottenuto usando la funzionalità generica PauliEvolutionGate().
t = Parameter("t")
# Create instruction for rotation about XX+YY-ZZ:
Rxyz_circ = QuantumCircuit(2)
Rxyz_circ.rxx(2 * JX * t, 0, 1)
Rxyz_circ.ryy(2 * JY * t, 0, 1)
Rxyz_circ.rzz(2 * JZ * t, 0, 1)
Rxyz_instr = Rxyz_circ.to_instruction(label="R J_x XX + J_y YY + J_z ZZ")
interaction_list = [
[[i, i + 1] for i in range(0, n_qubits - 1, 2)],
[[i, i + 1] for i in range(1, n_qubits - 1, 2)],
] # linear chain
qr = QuantumRegister(n_qubits)
trotter_step_circ = QuantumCircuit(qr)
for i, color in enumerate(interaction_list):
for interaction in color:
trotter_step_circ.append(Rxyz_instr, interaction)
if i < len(interaction_list) - 1:
trotter_step_circ.barrier()
reverse_trotter_step_circ = trotter_step_circ.reverse_ops()
qc_evol = QuantumCircuit(qr)
for step in range(num_trotter_steps):
if step % 2 == 0:
qc_evol = qc_evol.compose(trotter_step_circ)
else:
qc_evol = qc_evol.compose(reverse_trotter_step_circ)
qc_evol.decompose().draw("mpl", fold=-1, scale=0.5)
Prepariamo nuovamente uno stato iniziale per questo test di Hadamard efficiente.
control = 0
excitation = int(n_qubits / 2) + 1
controlled_state_prep = QuantumCircuit(n_qubits + 1)
controlled_state_prep.cx(control, excitation)
controlled_state_prep.draw("mpl", fold=-1, scale=0.5)

Circuiti template per il calcolo degli elementi di matrice di e tramite il test di Hadamard
L'unica differenza tra i circuiti usati nel test di Hadamard sarà la fase nell'operatore di evoluzione temporale e gli osservabili misurati. Possiamo quindi preparare un circuito template che rappresenta il circuito generico per il test di Hadamard, con segnaposto per i gate che dipendono dall'operatore di evoluzione temporale.
# Parameters for the template circuits
parameters = []
for idx in range(1, krylov_dim):
parameters.append(dt_circ * (idx))
# Create modified hadamard test circuit
qr = QuantumRegister(n_qubits + 1)
qc = QuantumCircuit(qr)
qc.h(0)
qc.compose(controlled_state_prep, list(range(n_qubits + 1)), inplace=True)
qc.barrier()
qc.compose(qc_evol, list(range(1, n_qubits + 1)), inplace=True)
qc.barrier()
qc.x(0)
qc.compose(controlled_state_prep.inverse(), list(range(n_qubits + 1)), inplace=True)
qc.x(0)
qc.decompose().draw("mpl", fold=-1)
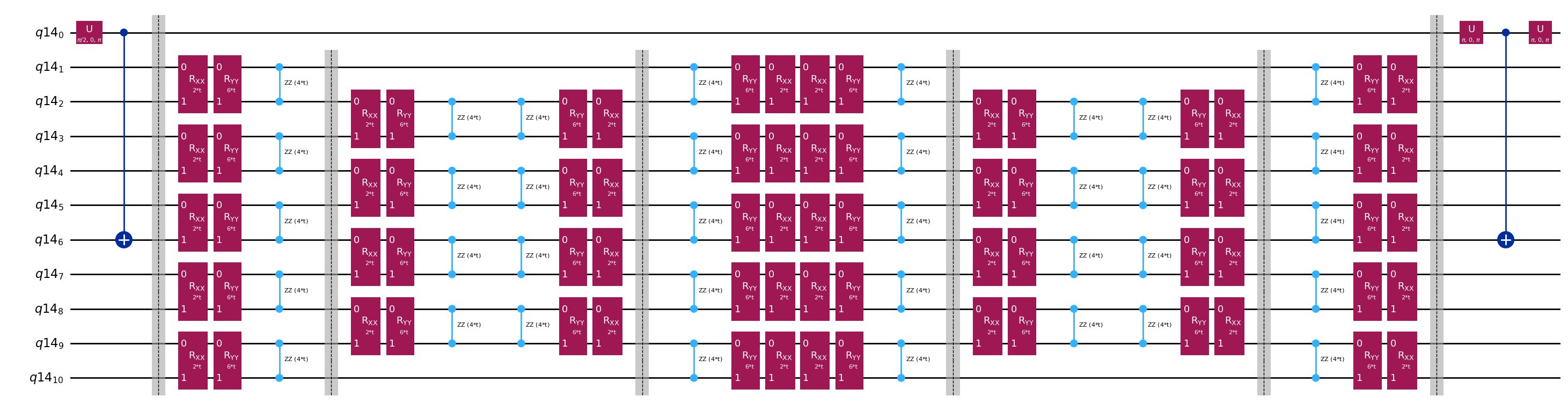
print(
"The optimized circuit has 2Q gates depth: ",
qc.decompose().decompose().depth(lambda x: x[0].num_qubits == 2),
)
The optimized circuit has 2Q gates depth: 50
Questa profondità è sostanzialmente ridotta rispetto al test di Hadamard originale. Questa profondità è gestibile sui computer quantistici moderni, sebbene sia ancora piuttosto elevata. Dovremo usare la mitigazione degli errori all'avanguardia per ottenere risultati utili.
Seleziona un backend su cui eseguire il calcolo quantistico di Krylov, in modo da poter transpilare il circuito per l'esecuzione su quel computer quantistico.
# Use the least-busy backend or specify a quantum computer using the syntax commented out below.
service = QiskitRuntimeService()
backend = service.least_busy(operational=True, simulator=False)
# Or you may choose a specify backend and channel if necessary for your workflow.
# service = QiskitRuntimeService(channel="ibm_quantum_platform")
# backend = service.backend("ibm_fez")
Ora transpiliamo i nostri circuiti e operatori.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
target = backend.target
basis_gates = list(target.operation_names)
pm = generate_preset_pass_manager(
optimization_level=3, backend=backend, basis_gates=basis_gates
)
qc_trans = pm.run(qc)
print(qc_trans.depth(lambda x: x[0].num_qubits == 2))
print(qc_trans.count_ops())
qc_trans.draw("mpl", fold=-1, idle_wires=False, scale=0.5)
36
OrderedDict([('rz', 410), ('sx', 361), ('cz', 156), ('x', 18), ('barrier', 6)])

Dopo l'ottimizzazione, la profondità a due qubit del circuito transpilato è ulteriormente ridotta.
4.3 Passo 3. Eseguire con una primitiva di Qiskit Runtime
Creiamo ora i PUB per l'esecuzione con Estimator.
# Define observables to measure for S
observable_S_real = "I" * (n_qubits) + "X"
observable_S_imag = "I" * (n_qubits) + "Y"
observable_op_real = SparsePauliOp(
observable_S_real
) # define a sparse pauli operator for the observable
observable_op_imag = SparsePauliOp(observable_S_imag)
layout = qc_trans.layout # get layout of transpiled circuit
observable_op_real = observable_op_real.apply_layout(
layout
) # apply physical layout to the observable
observable_op_imag = observable_op_imag.apply_layout(layout)
observable_S_real = (
observable_op_real.paulis.to_labels()
) # get the label of the physical observable
observable_S_imag = observable_op_imag.paulis.to_labels()
observables_S = [[observable_S_real], [observable_S_imag]]
# Define observables to measure for H
# Hamiltonian terms to measure
observable_list = []
for pauli, coeff in zip(H_op.paulis, H_op.coeffs):
# print(pauli)
observable_H_real = pauli[::-1].to_label() + "X"
observable_H_imag = pauli[::-1].to_label() + "Y"
observable_list.append([observable_H_real])
observable_list.append([observable_H_imag])
layout = qc_trans.layout
observable_trans_list = []
for observable in observable_list:
observable_op = SparsePauliOp(observable)
observable_op = observable_op.apply_layout(layout)
observable_trans_list.append([observable_op.paulis.to_labels()])
observables_H = observable_trans_list
# Define a sweep over parameter values
params = np.vstack(parameters).T
# Estimate the expectation value for all combinations of
# observables and parameter values, where the pub result will have
# shape (# observables, # parameter values).
pub = (qc_trans, observables_S + observables_H, params)
I circuiti per sono calcolabili classicamente. Lo eseguiamo prima di passare al caso usando un computer quantistico.
from qiskit.quantum_info import StabilizerState, Pauli
qc_cliff = qc.assign_parameters({t: 0})
# Get expectation values from experiment
S_expval_real = StabilizerState(qc_cliff).expectation_value(
Pauli("I" * (n_qubits) + "X")
)
S_expval_imag = StabilizerState(qc_cliff).expectation_value(
Pauli("I" * (n_qubits) + "Y")
)
# Get expectation values
S_expval = S_expval_real + 1j * S_expval_imag
H_expval = 0
for obs_idx, (pauli, coeff) in enumerate(zip(H_op.paulis, H_op.coeffs)):
# Get expectation values from experiment
expval_real = StabilizerState(qc_cliff).expectation_value(
Pauli(pauli[::-1].to_label() + "X")
)
expval_imag = StabilizerState(qc_cliff).expectation_value(
Pauli(pauli[::-1].to_label() + "Y")
)
expval = expval_real + 1j * expval_imag
# Fill-in matrix elements
H_expval += coeff * expval
print(H_expval)
(10+0j)
Sebbene siamo riusciti a ridurre la profondità del gate di ordini di grandezza usando il test di Hadamard efficiente, la profondità è ancora sufficiente da richiedere la mitigazione degli errori all'avanguardia. Di seguito specifichiamo gli attributi della mitigazione utilizzata. Tutti i metodi usati sono importanti, ma vale la pena sottolineare in particolare l'amplificazione probabilistica degli errori (PEA). Questa potente tecnica comporta un notevole overhead quantistico. Il calcolo eseguito qui può richiedere 20 minuti o più su un computer quantistico reale. Puoi modificare i parametri seguenti per aumentare o diminuire la precisione e di conseguenza l'overhead. Le impostazioni predefinite riportate di seguito producono risultati ad alta fedeltà.
# Experiment options
num_randomizations = 300
num_randomizations_learning = 20
max_batch_circuits = 20
shots_per_randomization = 100
learning_pair_depths = [0, 4, 24]
noise_factors = [1, 1.3, 1.6]
# Base option formatting
options = {
# Builtin resilience settings for ZNE
"resilience": {
"measure_mitigation": True,
"zne_mitigation": True,
"zne": {"noise_factors": noise_factors},
# TREX noise learning configuration
"measure_noise_learning": {
"num_randomizations": num_randomizations_learning,
"shots_per_randomization": shots_per_randomization,
},
# PEA noise model configuration
"layer_noise_learning": {
"max_layers_to_learn": 10,
"layer_pair_depths": learning_pair_depths,
"shots_per_randomization": shots_per_randomization,
"num_randomizations": num_randomizations_learning,
},
},
# Randomization configuration
"twirling": {
"num_randomizations": num_randomizations,
"shots_per_randomization": shots_per_randomization,
"strategy": "all",
},
# Experimental settings for PEA method
"experimental": {
# # Just in case, disable any further qiskit transpilation not related to twirling / DD
# "skip_transpilation": True,
# Execution configuration
"execution": {
"max_pubs_per_batch_job": max_batch_circuits,
"fast_parametric_update": True,
},
# Error Mitigation configuration
"resilience": {
# ZNE Configuration
"zne": {
"amplifier": "pea",
"return_all_extrapolated": True,
"return_unextrapolated": True,
"extrapolated_noise_factors": [0] + noise_factors,
}
},
},
}
Infine, eseguiamo i circuiti per e con Estimator.
# This job required 17 minutes of QPU time to run on a Heron r2 processor. This is only an estimate.
# Your execution time may vary.
with Batch(backend=backend) as batch:
# Estimator
estimator = Estimator(mode=batch, options=options)
job = estimator.run([pub], precision=1)
4.4 Passo 4. Post-elaborazione e analisi dei risultati
Quello che abbiamo ottenuto dal computer quantistico sono i singoli elementi di matrice di e i gruppi di Pauli commutanti che compongono gli elementi di matrice di . Questi termini devono essere combinati per recuperare le nostre matrici, in modo da poter risolvere il problema agli autovalori generalizzato.
# Store the outputs as 'results'.
results = job.result()[0]
Calcola l'Hamiltoniano effettivo e le matrici di sovrapposizione
Prima calcola la fase accumulata dallo stato durante l'evoluzione temporale non controllata
prefactors = [
np.exp(-1j * sum([c for p, c in H_op.to_list() if "Z" in p]) * i * dt)
for i in range(1, krylov_dim)
]
Una volta ottenuti i risultati delle esecuzioni dei circuiti, possiamo post-elaborare i dati per calcolare gli elementi di matrice di
# Assemble S, the overlap matrix of dimension D:
S_first_row = np.zeros(krylov_dim, dtype=complex)
S_first_row[0] = 1 + 0j
# Add in ancilla-only measurements:
for i in range(krylov_dim - 1):
# Get expectation values from experiment
expval_real = results.data.evs[0][0][i] # automatic extrapolated evs if ZNE is used
expval_imag = results.data.evs[1][0][i] # automatic extrapolated evs if ZNE is used
# Get expectation values
expval = expval_real + 1j * expval_imag
S_first_row[i + 1] += prefactors[i] * expval
S_first_row_list = S_first_row.tolist() # for saving purposes
S_circ = np.zeros((krylov_dim, krylov_dim), dtype=complex)
# Distribute entries from first row across matrix:
for i, j in it.product(range(krylov_dim), repeat=2):
if i >= j:
S_circ[j, i] = S_first_row[i - j]
else:
S_circ[j, i] = np.conj(S_first_row[j - i])
from sympy import Matrix
Matrix(S_circ)
E gli elementi di matrice di
import itertools
# Assemble S, the overlap matrix of dimension D:
H_first_row = np.zeros(krylov_dim, dtype=complex)
H_first_row[0] = H_expval
for obs_idx, (pauli, coeff) in enumerate(zip(H_op.paulis, H_op.coeffs)):
# Add in ancilla-only measurements:
for i in range(krylov_dim - 1):
# Get expectation values from experiment
expval_real = results.data.evs[2 + 2 * obs_idx][0][
i
] # automatic extrapolated evs if ZNE is used
expval_imag = results.data.evs[2 + 2 * obs_idx + 1][0][
i
] # automatic extrapolated evs if ZNE is used
# Get expectation values
expval = expval_real + 1j * expval_imag
H_first_row[i + 1] += prefactors[i] * coeff * expval
H_first_row_list = H_first_row.tolist()
H_eff_circ = np.zeros((krylov_dim, krylov_dim), dtype=complex)
# Distribute entries from first row across matrix:
for i, j in itertools.product(range(krylov_dim), repeat=2):
if i >= j:
H_eff_circ[j, i] = H_first_row[i - j]
else:
H_eff_circ[j, i] = np.conj(H_first_row[j - i])
from sympy import Matrix
Matrix(H_eff_circ)
Infine, possiamo risolvere il problema agli autovalori generalizzato per :
e ottenere una stima dell'energia dello stato fondamentale
gnd_en_circ_est_list = []
for d in range(1, krylov_dim + 1):
# Solve generalized eigenvalue problem
gnd_en_circ_est = solve_regularized_gen_eig(
H_eff_circ[:d, :d], S_circ[:d, :d], threshold=1e-1
)
gnd_en_circ_est_list.append(gnd_en_circ_est)
print("The estimated ground state energy is: ", gnd_en_circ_est)
The estimated ground state energy is: 10.0
The estimated ground state energy is: 5.933953916292923
The estimated ground state energy is: 4.4101773995740645
The estimated ground state energy is: 3.921288588521255
Per un settore a singola particella, possiamo calcolare in modo efficiente classicamente lo stato fondamentale di questo settore dell'Hamiltoniano
gs_en = single_particle_gs(H_op, n_qubits)
n_sys_qubits 10
n_exc 1 , subspace dimension 11
single particle ground state energy: 2.391547869638771
len(H_op)
27
plt.plot(
range(1, krylov_dim + 1),
gnd_en_circ_est_list,
color="blue",
linestyle="-.",
label="KQD estimate",
)
plt.plot(
range(1, krylov_dim + 1),
[gs_en] * krylov_dim,
color="red",
linestyle="-",
label="exact",
)
plt.xticks(range(1, krylov_dim + 1), range(1, krylov_dim + 1))
plt.legend()
plt.xlabel("Krylov space dimension")
plt.ylabel("Energy")
plt.title("Estimating Ground state energy with Krylov Quantum Diagonalization")
plt.show()
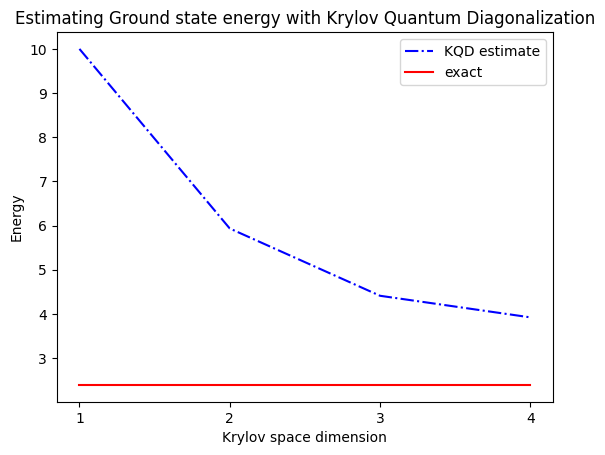
5. Discussione ed estensioni
Per ricapitolare: partiamo da uno stato di riferimento, poi lo facciamo evolvere per diversi intervalli di tempo al fine di generare il sottospazio di Krylov unitario. Proiettiamo il nostro Hamiltoniano su quel sottospazio. Stimiamo anche le sovrapposizioni dei vettori del sottospazio. Infine risolviamo classicamente il problema agli autovalori generalizzato di dimensione ridotta.
Confrontiamo cosa determina i costi computazionali dell'utilizzo della tecnica di Krylov in modo classico e quantistico. Non esistono analoghi perfetti tra l'approccio classico e quello quantistico per tutti i passaggi. Questa tabella raccoglie alcune scalature di diversi passaggi a titolo di confronto.
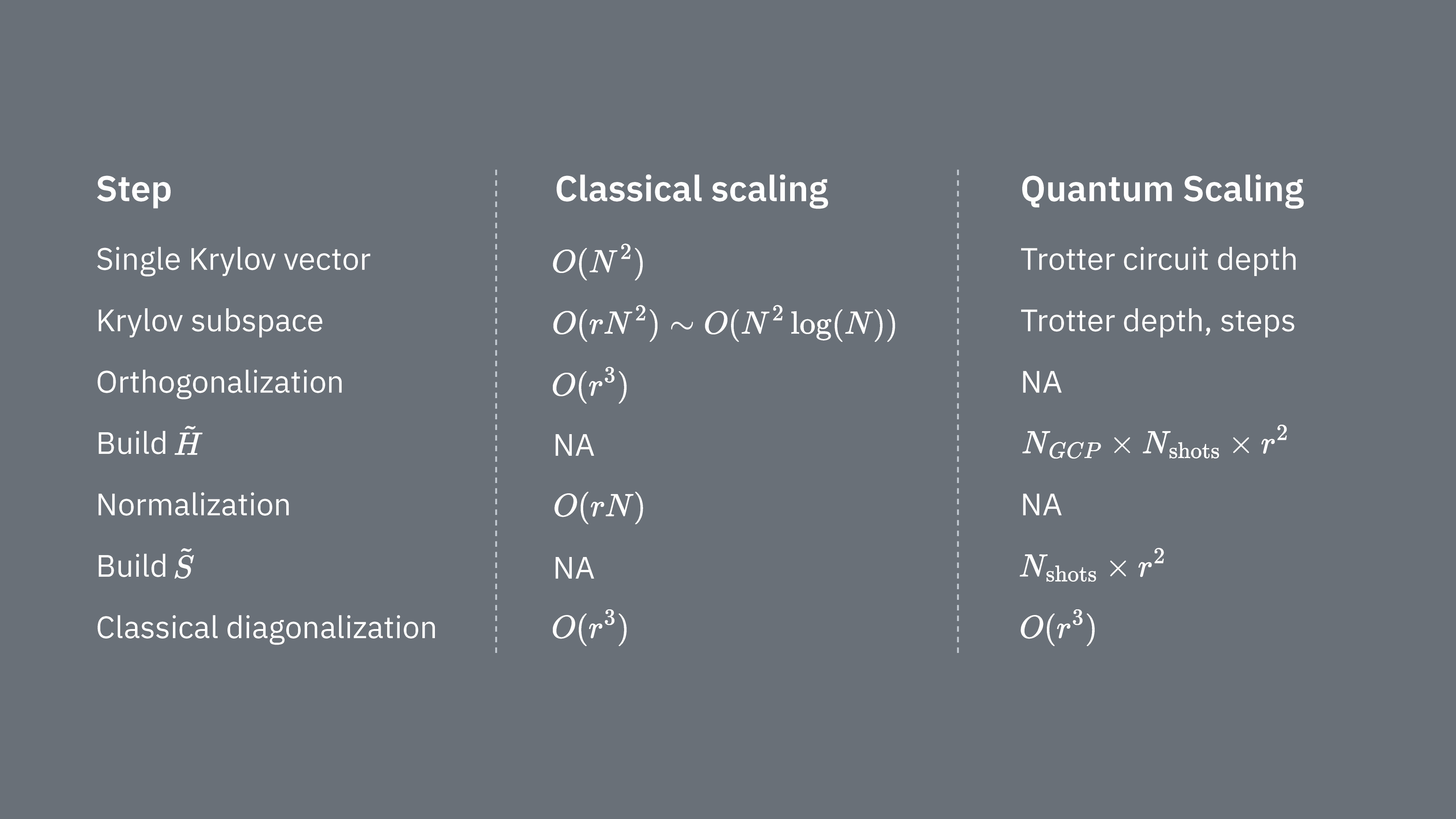
Ricorda che gli Hamiltoniani hanno in generale termini che non possono essere misurati simultaneamente (perché non commutano tra loro). Raggruppiamo i termini dell'Hamiltoniano in insiemi di operatori di Pauli commutanti che possono essere tutti misurati contemporaneamente; potrebbero essere necessari molti di questi gruppi per tenere conto di tutti i termini che non commutano tra loro. Per costruire su un computer quantistico sono necessarie misurazioni separate per ciascun gruppo di stringhe di Pauli commutanti nell'Hamiltoniano, e ognuna di esse richiede molti shot. Dobbiamo farlo per elementi di matrice diversi, corrispondenti a combinazioni di diversi fattori di evoluzione temporale. Esistono talvolta modi per ridurre questo numero, ma in questa trattazione approssimativa il tempo richiesto scala come Gli elementi di devono essere stimati, il che scala come . Infine, risolvere classicamente il problema agli autovalori generalizzato nello spazio proiettato richiede
Vediamo che la diagonalizzazione quantistica di Krylov può essere utile nei casi in cui il numero di gruppi di Pauli commutanti nell'Hamiltoniano è relativamente piccolo. Queste dipendenze di scalatura suggeriscono alcune applicazioni in cui il metodo di Krylov può essere utile, e altre in cui probabilmente non lo sarà. Alcuni Hamiltoniani hanno un'alta complessità quando vengono mappati su qubit, coinvolgendo molte stringhe di Pauli non commutanti che non possono essere facilmente suddivise in pochi gruppi commutanti. Ciò è spesso vero, ad esempio, per i problemi di chimica quantistica. Questa complessità presenta due sfide principali per i computer quantistici nel breve termine:
- La stima di ogni elemento di diventa computazionalmente onerosa a causa dell'elevato numero di termini.
- I circuiti di Trotter richiesti diventano troppo profondi.
Entrambi i punti precedenti saranno meno problematici quando i computer quantistici raggiungeranno la tolleranza agli errori, ma devono essere considerati nel breve termine. Anche i sistemi con mappature "più semplici" di quelle della chimica quantistica possono incorrere negli stessi impedimenti, se gli Hamiltoniani hanno troppi termini non commutanti. Il metodo di Krylov è più utile quando l'Hamiltoniano può essere suddiviso in relativamente pochi gruppi di Pauli commutanti e quando è facile da implementare nei circuiti di Trotter. Entrambe queste condizioni sono soddisfatte, ad esempio, per molti modelli su reticolo di interesse in fisica. KQD è particolarmente utile quando si sa molto poco dello stato fondamentale. Ciò deriva dalle sue garanzie di convergenza intrinseche e dalla sua applicabilità in scenari in cui metodi alternativi non sono praticabili per insufficiente conoscenza dello stato fondamentale.
Sebbene KQD sia uno strumento potente, gli aspetti più dispendiosi in termini di tempo del protocollo — in particolare la stima di ogni elemento dell'Hamiltoniano proiettato e della sovrapposizione degli stati di Krylov — rappresentano opportunità di miglioramento. Un approccio alternativo prevede di sfruttare i metodi di Krylov in combinazione con metodi basati sul campionamento, che sono l'oggetto della lezione successiva.
6. Appendici
Appendice I: sottospazio di Krylov da evoluzioni temporali reali
Lo spazio di Krylov unitario è definito come
per un certo passo temporale che determineremo in seguito. Supponiamo temporaneamente che sia pari: definiamo allora . Nota che quando proiettiamo l'Hamiltoniano nel sottospazio di Krylov sopra, esso è indistinguibile dal sottospazio di Krylov
ovvero quello in cui tutte le evoluzioni temporali sono traslate indietro di passi temporali. Il motivo per cui è indistinguibile è che gli elementi di matrice
sono invarianti rispetto a traslazioni globali del tempo di evoluzione, poiché le evoluzioni temporali commutano con l'Hamiltoniano. Per dispari, si può usare l'analisi per .
Vogliamo dimostrare che da qualche parte in questo sottospazio di Krylov è garantita la presenza di uno stato a bassa energia. Lo facciamo mediante il seguente risultato, derivato dal Teorema 3.1 in [3]:
Affermazione 1: esiste una funzione tale che per le energie nell'intervallo spettrale dell'Hamiltoniano (cioè tra l'energia dello stato fondamentale e l'energia massima)...
- per tutti i valori di che si trovano a distanza da , ossia è soppressa esponenzialmente
- è una combinazione lineare di per
Di seguito forniamo una dimostrazione, che può essere tranquillamente saltata a meno che non si voglia comprendere l'argomento completo e rigoroso. Per ora ci concentriamo sulle implicazioni dell'affermazione precedente. Dalla proprietà 3, possiamo vedere che il sottospazio di Krylov traslato sopra contiene lo stato . Questo è il nostro stato a bassa energia. Per capire perché, scriviamo nella base degli autostati energetici:
dove è il k-esimo autostato energetico e è la sua ampiezza nello stato iniziale . Espresso in questi termini, è dato da
usando il fatto che possiamo sostituire con quando agisce sull'autostato . L'errore energetico di questo stato è quindi
Per trasformare questo in un limite superiore più facile da comprendere, separiamo prima la somma al numeratore nei termini con e nei termini con :
Possiamo limitare superiormente il primo termine con ,
dove il primo passo segue perché per ogni nella somma, e il secondo passo segue perché la somma al numeratore è un sottoinsieme della somma al denominatore. Per il secondo termine, limitiamo inferiormente il denominatore con , poiché : sommando tutto di nuovo insieme, si ottiene
Per semplificare ciò che rimane, nota che per tutti questi , dalla definizione di sappiamo che . Limitando ulteriormente e si ottiene
Questo vale per qualsiasi ; se impostiamo uguale all'errore obiettivo, il limite dell'errore converge verso di esso esponenzialmente con la dimensione di Krylov . Nota anche che se , il termine nel limite precedente scompare del tutto.
Per completare l'argomento, osserviamo innanzitutto che quanto sopra è solo l'errore energetico del particolare stato , non l'errore energetico dello stato a energia più bassa nel sottospazio di Krylov. Tuttavia, per il principio variazionale (Rayleigh-Ritz), l'errore energetico dello stato a energia più bassa nel sottospazio di Krylov è limitato superiormente dall'errore energetico di qualsiasi stato nel sottospazio, quindi quanto sopra è anche un limite superiore all'errore energetico dello stato a energia più bassa, cioè l'output dell'algoritmo di diagonalizzazione quantistica di Krylov.
Un'analisi simile a quella precedente può essere condotta tenendo conto anche del rumore e della procedura di sogliatura discussa nel notebook. Si vedano [2] e [4] per questa analisi.
Appendice II: dimostrazione dell'Affermazione 1
Il seguente risultato è principalmente derivato da [3], Teorema 3.1: siano e sia lo spazio dei polinomi residui (polinomi il cui valore in 0 è 1) di grado al più . La soluzione di
è
e il corrispondente valore minimo è
Vogliamo convertire questo in una funzione che possa essere espressa naturalmente in termini di esponenziali complessi, poiché queste sono le evoluzioni temporali reali che generano il sottospazio di Krylov quantistico. A tale scopo, è utile introdurre la seguente trasformazione delle energie nell'intervallo spettrale dell'Hamiltoniano in numeri nell'intervallo : definiamo
dove è un passo temporale tale che . Nota che e cresce man mano che si allontana da .
Ora, usando il polinomio con i parametri a, b, d impostati su , e d = int(r/2), definiamo la funzione:
dove è l'energia dello stato fondamentale. Inserendo si può vedere che è un polinomio trigonometrico di grado , ossia una combinazione lineare di per . Inoltre, dalla definizione di abbiamo che e per qualsiasi nell'intervallo spettrale tale che abbiamo
Riferimenti:
[1] https://arxiv.org/abs/2407.14431
[2] https://arxiv.org/abs/1811.09025
[3] https://people.math.ethz.ch/~mhg/pub/biksm.pdf
[4] https://academic.oup.com/book/36426
[5] https://en.wikipedia.org/wiki/Krylov_subspace
[6] Krylov Subspace Methods: Principles and Analysis, Jörg Liesen, Zdenek Strakos https://academic.oup.com/book/36426
[7] Iterative Methods for Sparse Linear Systems" by Yousef Saad
[8] "MINRES-QLP: A Krylov Subspace Method for Indefinite or Singular Symmetric Systems" by Sou-Cheng Choi, Christopher Paige, and Michael Saunders (https://epubs.siam.org/doi/10.1137/100787921)
[9] Ethan N. Epperly, Lin Lin, and Yuji Nakatsukasa. "A theory of quantum subspace diagonalization". SIAM Journal on Matrix Analysis and Applications 43, 1263–1290 (2022).