Il variational quantum eigensolver (VQE)
Questa lezione presenta il variational quantum eigensolver, ne illustra l'importanza come algoritmo fondamentale nel quantum computing e ne esplora punti di forza e debolezze. Il VQE da solo, senza metodi di potenziamento, difficilmente sarà sufficiente per computazioni quantistiche alla scala dell'utilità moderna. Rimane tuttavia importante come metodo ibrido classico-quantistico archetipico, ed è una base fondamentale su cui sono costruiti molti algoritmi più avanzati.
Questo video offre una panoramica del VQE e dei fattori che ne influenzano l'efficienza. Il testo seguente aggiunge ulteriori dettagli e implementa il VQE usando Qiskit.
1. Cos'è il VQE?
Il variational quantum eigensolver è un algoritmo che utilizza il calcolo classico e quello quantistico in combinazione per portare a termine un compito. Un calcolo VQE è composto da quattro componenti principali:
- Un operatore: spesso un Hamiltoniano, che chiameremo , che descrive una proprietà del sistema che si desidera ottimizzare. In altri termini, si cerca l'autovettore di questo operatore corrispondente all'autovalore minimo. Quell'autovettore viene spesso chiamato "stato fondamentale".
- Un "ansatz" (parola tedesca che significa "approccio"): è un circuito quantistico che prepara uno stato quantistico che approssima l'autovettore cercato. In realtà l'ansatz è una famiglia di circuiti quantistici, perché alcuni dei gate dell'ansatz sono parametrizzati, cioè ricevono in ingresso un parametro che può essere variato. Questa famiglia di circuiti quantistici può preparare una famiglia di stati quantistici che approssimano lo stato fondamentale.
- Un Estimator: un mezzo per stimare il valore di aspettazione dell'operatore nello stato quantistico variazionale corrente. A volte ciò che interessa è semplicemente questo valore di aspettazione, che chiamiamo funzione di costo. A volte ci interessa una funzione più complessa che può comunque essere espressa a partire da uno o più valori di aspettazione.
- Un ottimizzatore classico: un algoritmo che varia i parametri per cercare di minimizzare la funzione di costo.
Esaminiamo ciascuno di questi componenti più in dettaglio.
1.1 L'operatore (Hamiltoniano)
Al centro di un problema VQE vi è un operatore che descrive un sistema di interesse. Si assume qui che l'autovalore minimo e il corrispondente autovettore di questo operatore siano utili per qualche scopo scientifico o applicativo. Esempi possono includere un Hamiltoniano chimico che descrive una molecola, in cui l'autovalore minimo dell'operatore corrisponde all'energia dello stato fondamentale della molecola e il corrispondente autostato ne descrive la geometria o la configurazione elettronica. Oppure l'operatore potrebbe descrivere il costo di un determinato processo da ottimizzare, e gli autostati potrebbero corrispondere a percorsi o procedure. In alcuni campi, come la fisica, un "Hamiltoniano" si riferisce quasi sempre a un operatore che descrive l'energia di un sistema fisico. Nel quantum computing, invece, è comune vedere operatori quantistici che descrivono un problema di business o logistico essere anch'essi denominati "Hamiltoniano". Adotteremo questa convenzione anche qui.

La mappatura di un problema fisico o di ottimizzazione sui qubit è tipicamente un compito non banale, ma questi dettagli non sono al centro di questo corso. Una discussione generale della mappatura di un problema su un operatore quantistico si trova in Quantum computing in practice. Un'analisi più approfondita della mappatura dei problemi di chimica su operatori quantistici si trova in Quantum Chemistry with VQE.
Ai fini di questo corso, assumeremo che la forma dell'Hamiltoniano sia nota. Ad esempio, un Hamiltoniano per una semplice molecola di idrogeno (sotto certe assunzioni sullo spazio attivo, e utilizzando il mapper Jordan-Wigner) è:
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-ibm-runtime scipy
from qiskit.quantum_info import SparsePauliOp
hamiltonian = SparsePauliOp(
[
"IIII",
"IIIZ",
"IZII",
"IIZI",
"ZIII",
"IZIZ",
"IIZZ",
"ZIIZ",
"IZZI",
"ZZII",
"ZIZI",
"YYYY",
"XXYY",
"YYXX",
"XXXX",
],
coeffs=[
-0.09820182 + 0.0j,
-0.1740751 + 0.0j,
-0.1740751 + 0.0j,
0.2242933 + 0.0j,
0.2242933 + 0.0j,
0.16891402 + 0.0j,
0.1210099 + 0.0j,
0.16631441 + 0.0j,
0.16631441 + 0.0j,
0.1210099 + 0.0j,
0.17504456 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
],
)
Nota che nell'Hamiltoniano sopra ci sono termini come ZZII e YYYY che non commutano tra loro. Cioè, per valutare ZZII, occorrerebbe misurare l'operatore di Pauli Z sul qubit 3 (tra le altre misurazioni). Ma per valutare YYYY, è necessario misurare l'operatore di Pauli Y su quello stesso qubit, il qubit 3. Tra gli operatori Y e Z sullo stesso qubit esiste una relazione di indeterminazione; non è possibile misurare entrambi gli operatori contemporaneamente. Torneremo su questo punto più avanti, e in effetti durante tutto il corso.
L'Hamiltoniano sopra è un operatore matriciale . Diagonalizzare l'operatore per trovare il suo autovalore energetico minimo non è difficile.
import numpy as np
A = np.array(hamiltonian)
eigenvalues, eigenvectors = np.linalg.eigh(A)
print("The ground state energy is ", min(eigenvalues), "hartrees")
The ground state energy is -1.1459778447627311 hartrees
I risolutori classici con forza bruta non riescono a scalare per descrivere le energie o le geometrie di sistemi molto grandi di atomi, come farmaci o proteine. Il VQE è uno dei primi tentativi di sfruttare il quantum computing in questo ambito.
In questa lezione incontreremo Hamiltoniani molto più grandi di quello precedente. Ma sarebbe prematuro spingere i limiti di ciò che il VQE può fare, prima di introdurre alcuni degli strumenti più avanzati che possono potenziarlo o sostituirlo, più avanti in questo corso.
1.2 Ansatz
La parola "ansatz" è tedesca e significa "approccio". Il plurale corretto in tedesco è "ansätze", benché si vedano spesso anche "ansatzes" o "ansatze". Nel contesto del VQE, un ansatz è il circuito quantistico che si usa per creare una funzione d'onda a più qubit che approssimi il più possibile lo stato fondamentale del sistema studiato, producendo così il valore di aspettazione minimo dell'operatore. Questo circuito quantistico conterrà parametri variazionali (spesso raccolti nel vettore di variabili ).

Viene scelto un insieme iniziale di valori dei parametri variazionali. Chiameremo l'operazione unitaria dell'ansatz sul circuito. Per impostazione predefinita, tutti i qubit nei computer quantistici IBM® vengono inizializzati allo stato . Quando il circuito viene eseguito, lo stato dei qubit sarà
Se ci bastasse ottenere l'energia minima (usando il linguaggio dei sistemi fisici), potremmo stimarla semplicemente misurando l'energia molte volte e prendendo il valore minimo. Tuttavia, in genere vogliamo anche la configurazione che produce quell'energia minima o autovalore. Il passo successivo è quindi la stima del valore di aspettazione dell'Hamiltoniano, che si ottiene tramite misurazioni quantistiche. Ci sono molti aspetti da considerare. Ma possiamo comprendere questo processo qualitativamente notando che la probabilità di misurare un'energia (usando ancora il linguaggio dei sistemi fisici) è correlata al valore di aspettazione da:
La probabilità è anche correlata alla sovrapposizione tra l'autostato e lo stato corrente del sistema :
Effettuando molte misurazioni degli operatori di Pauli che compongono il nostro Hamiltoniano, possiamo quindi stimare il valore di aspettazione dell'Hamiltoniano nello stato corrente del sistema . Il passo successivo è variare i parametri per avvicinarci il più possibile allo stato fondamentale (a energia minima) del sistema. Per via dei parametri variazionali nell'ansatz, a volte quest'ultimo viene chiamato forma variazionale.
Prima di passare al processo variazionale, è utile notare che conviene spesso iniziare da uno stato "di buona approssimazione iniziale". Se si sa abbastanza del proprio sistema, si può fare una supposizione iniziale migliore di . Per esempio, nelle applicazioni chimiche è comune inizializzare i qubit nello stato di Hartree-Fock. Questa ipotesi di partenza, che non contiene parametri variazionali, è chiamata stato di riferimento. Chiamiamo il circuito quantistico usato per creare lo stato di riferimento. Ogni volta che diventa importante distinguere lo stato di riferimento dal resto dell'ansatz, si usa: In modo equivalente
1.3 Stimatore
Abbiamo bisogno di un modo per stimare il valore di aspettazione del nostro Hamiltoniano in un particolare stato variazionale . Se potessimo misurare direttamente l'intero operatore , basterebbe effettuare molte (diciamo ) misurazioni e calcolarne la media:
Qui il simbolo ci ricorda che questo valore di aspettazione sarebbe esatto solo nel limite . Ma con migliaia di misurazioni effettuate su un circuito, l'errore di campionamento del valore di aspettazione è piuttosto basso. Esistono altre considerazioni, come il rumore, che diventano rilevanti per calcoli molto precisi.
In generale, però, non è possibile misurare tutto in una volta. può contenere più operatori di Pauli X, Y e Z non commutanti. L'Hamiltoniano deve quindi essere suddiviso in gruppi di operatori che possono essere misurati simultaneamente; ciascun gruppo deve essere stimato separatamente e i risultati combinati per ottenere un valore di aspettazione. Torneremo su questo aspetto in modo più dettagliato nella prossima lezione, quando discuteremo la scalabilità degli approcci classici e quantistici. Questa complessità nella misurazione è uno dei motivi per cui abbiamo bisogno di codice altamente efficiente per effettuare tali stime. In questa lezione e oltre, utilizzeremo la primitiva Estimator di Qiskit Runtime a tale scopo.
1.4 Ottimizzatori classici
Un ottimizzatore classico è qualsiasi algoritmo classico progettato per trovare gli estremi di una funzione target (tipicamente un minimo). Esplora lo spazio dei parametri possibili alla ricerca di un insieme che minimizzi una funzione di interesse. Possono essere suddivisi in linea di massima in metodi basati sul gradiente, che utilizzano informazioni sul gradiente, e metodi privi di gradiente, che operano come ottimizzatori a scatola nera. La scelta dell'ottimizzatore classico può influenzare significativamente le prestazioni dell'algoritmo, specialmente in presenza di rumore nell'hardware quantistico. Tra gli ottimizzatori più diffusi in questo campo vi sono Adam, AMSGrad e SPSA, che hanno mostrato risultati promettenti in ambienti rumorosi. Tra gli ottimizzatori più tradizionali ci sono COBYLA e SLSQP.
Un flusso di lavoro comune (dimostrato nella Sezione 3.3) consiste nell'usare uno di questi algoritmi come metodo all'interno di un minimizzatore come la funzione minimize di scipy. Questa accetta come argomenti:
- Una funzione da minimizzare. Spesso è il valore di aspettazione dell'energia. Ma in generale si chiamano "funzioni di costo".
- Un insieme di parametri da cui iniziare la ricerca. Spesso chiamati o .
- Argomenti, inclusi gli argomenti della funzione di costo. Nel quantum computing con Qiskit, questi argomenti includeranno l'ansatz, l'Hamiltoniano e la primitiva Estimator, di cui si parla più nella prossima sottosezione.
- Un 'metodo' di minimizzazione. Questo si riferisce all'algoritmo specifico usato per esplorare lo spazio dei parametri. È qui che si specifica, per esempio, COBYLA o SLSQP.
- Opzioni. Le opzioni disponibili possono variare in base al metodo. Ma un esempio che quasi tutti i metodi includono è il numero massimo di iterazioni dell'ottimizzatore prima di terminare la ricerca: 'maxiter'.

Ad ogni passo iterativo, il valore di aspettazione dell'Hamiltoniano viene stimato effettuando molte misurazioni. Questa energia stimata viene restituita dalla funzione di costo, e il minimizzatore aggiorna le informazioni che possiede sul paesaggio energetico. Ciò che l'ottimizzatore fa esattamente per scegliere il passo successivo varia da metodo a metodo. Alcuni usano i gradienti e selezionano la direzione di discesa più ripida. Altri possono tenere conto del rumore e potrebbero richiedere che il costo diminuisca di un margine significativo prima di accettare che l'energia reale diminuisca in quella direzione.
# Example syntax for minimization
# from scipy.optimize import minimize
# res = minimize(cost_func, x0, args=(ansatz, hamiltonian, estimator), method="cobyla",
# options={'maxiter': 200})
1.5 Il principio variazionale
In questo contesto il principio variazionale è molto importante; esso afferma che nessuna funzione d'onda variazionale può produrre un valore di aspettazione dell'energia (o del costo) inferiore a quello prodotto dalla funzione d'onda dello stato fondamentale. Matematicamente,
Questo è facile da verificare se si nota che l'insieme di tutti gli autostati di forma una base completa per lo spazio di Hilbert. In altre parole, qualsiasi stato e in particolare può essere scritto come una somma pesata (normalizzata) di questi autostati di :
dove sono costanti da determinare, e . Lasciamo questo come esercizio al lettore. Ma si noti l'implicazione: lo stato variazionale che produce il valore di aspettazione dell'energia più basso è la migliore stima del vero stato fondamentale.
Verifica la tua comprensione
Verifica matematicamente che per qualsiasi stato variazionale .
Risposta
Usando la data espansione dello stato variazionale in termini degli autostati dell'energia,
possiamo scrivere il valore di aspettazione dell'energia variazionale come
Per tutti i coefficienti . Quindi possiamo scrivere
2. Confronto con il flusso di lavoro classico
Supponiamo di essere interessati a una matrice con N righe e N colonne. Supponi che la tua matrice sia così grande che la diagonalizzazione esatta non sia un'opzione. Supponi inoltre di sapere abbastanza del tuo problema da poter fare alcune ipotesi sulla struttura complessiva dell'autostato target, e di voler esplorare stati simili alla tua ipotesi iniziale per vedere se il costo/energia può essere ulteriormente ridotto. Questo è un approccio variazionale, ed è uno dei metodi utilizzati quando la diagonalizzazione esatta non è un'opzione.
2.1 Flusso di lavoro classico
Usando un computer classico, il processo funzionerebbe così:
- Fai un'ipotesi sullo stato, con alcuni parametri che varierai: . Sebbene questa ipotesi iniziale potrebbe essere casuale, non è consigliabile. Vogliamo usare la conoscenza del problema in questione per personalizzare la nostra ipotesi il più possibile.
- Calcola il valore di aspettazione dell'operatore con il sistema in quello stato:
- Modifica i parametri variazionali e ripeti: .
- Usa le informazioni accumulate sul paesaggio degli stati possibili nel tuo sottospazio variazionale per fare ipotesi sempre migliori e avvicinarti allo stato target. Il principio variazionale garantisce che il nostro stato variazionale non possa produrre un autovalore inferiore a quello dello stato fondamentale target. Quindi più basso è il valore di aspettazione, migliore è la nostra approssimazione dello stato fondamentale:
Esaminiamo la difficoltà di ogni passo in questo approccio. Impostare o aggiornare i parametri è computazionalmente semplice; la difficoltà sta nel selezionare parametri iniziali utili e fisicamente motivati. Usare le informazioni accumulate dalle iterazioni precedenti per aggiornare i parametri in modo da avvicinarsi allo stato fondamentale è non banale. Ma esistono algoritmi di ottimizzazione classici che lo fanno in modo piuttosto efficiente. Questa ottimizzazione classica è costosa solo perché potrebbe richiedere molte iterazioni; nel caso peggiore, il numero di iterazioni può scalare in modo esponenziale con N. Il singolo passo computazionalmente più oneroso è quasi certamente il calcolo del valore di aspettazione della matrice usando uno stato dato :
La matrice deve agire sul vettore di elementi, il che corrisponde a: operazioni di moltiplicazione nel caso peggiore. Questo deve essere fatto ad ogni iterazione dei parametri. Per matrici estremamente grandi, questo ha un costo computazionale elevato.
2.2 Flusso di lavoro quantistico e gruppi di Pauli commutanti
Immagina ora di affidare questa parte del calcolo a un computer quantistico. Invece di calcolare questo valore di aspettazione, lo stimi preparando lo stato sul computer quantistico tramite il tuo ansatz variazionale, e poi effettuando misurazioni.
Questo può sembrare più semplice di quanto sia in realtà. in generale non è facile da misurare. Potrebbe ad esempio essere composto da molti operatori di Pauli X, Y e Z non commutanti. Ma può essere scritto come combinazione lineare di termini , ognuno dei quali è facilmente misurabile (per esempio, operatori di Pauli o gruppi di operatori di Pauli che commutano qubit per qubit). Il valore di aspettazione di in uno stato è la somma pesata dei valori di aspettazione dei termini costitutivi . Questa espressione vale per qualsiasi stato , ma la useremo specificamente con i nostri stati variazionali .
dove è una stringa di Pauli come IZZX…XIYX, o più stringhe di questo tipo che commutano tra loro. Quindi una descrizione del valore di aspettazione che si avvicina maggiormente alla realtà delle misurazioni sui computer quantistici è
E nel contesto della nostra funzione d'onda variazionale:
Ciascuno dei termini può essere misurato volte producendo campioni di misura con e restituendo un valore di aspettazione e una deviazione standard . Si possono sommare questi termini e propagare gli errori attraverso la somma per ottenere un valore di aspettazione complessivo e una deviazione standard .
Questo non richiede moltiplicazioni su larga scala, né alcun processo che scala necessariamente come . Richiede invece molteplici misurazioni sul computer quantistico. Se non ne servono troppe, questo approccio può essere efficiente. Ed è questa la parte quantistica del VQE.
Ma parliamo dei motivi per cui potrebbe non essere efficiente. Un motivo per molte misurazioni è ridurre l'incertezza statistica nelle stime, per calcoli di altissima precisione. Un altro motivo è il numero di stringhe di Pauli necessarie per rappresentare l'intera matrice. Poiché le matrici di Pauli (più l'identità: X, Y, Z e I) spannano lo spazio di tutti gli operatori di una data dimensione, siamo garantiti di poter scrivere la nostra matrice di interesse come somma pesata di operatori di Pauli, come fatto in precedenza.
dove è una stringa di Pauli che agisce su tutti i qubit che descrivono il tuo sistema come IZZX…XIYX, o più stringhe di questo tipo che commutano tra loro. Ricorda che Qiskit usa la notazione little endian, in cui l'-esimo operatore di Pauli da destra agisce sull'-esimo qubit. Quindi possiamo misurare il nostro operatore misurando una serie di operatori di Pauli.
Ma non possiamo misurare tutti questi operatori di Pauli contemporaneamente. Gli operatori di Pauli (escluso I) non commutano tra loro se sono associati allo stesso qubit. Per esempio, possiamo misurare IZIZ e ZZXZ simultaneamente, perché possiamo misurare I e Z contemporaneamente per il terzo qubit, e possiamo conoscere I e X contemporaneamente per il primo qubit. Ma non possiamo misurare ZZZZ e ZZZX simultaneamente, perché Z e X non commutano, e agiscono entrambi sul qubit 0. I lettori esperti potrebbero ricordare che due gruppi di operatori di Pauli possono commutare come insieme anche se le misurazioni dei singoli qubit non commutano. Estimator assume misurazioni di Pauli come prodotto tensoriale (tramite rotazioni della base), corrispondenti al raggruppamento di operatori che commutano qubit per qubit. Quindi, per stimare contemporaneamente due stringhe (A e B) di operatori di Pauli usando Estimator, gli operatori di Pauli di ogni qubit in A e B devono commutare. Ciò significa che non possiamo misurare nemmeno ZZZZ e ZZXX simultaneamente.

Quindi decomponiamo la nostra matrice in una somma di Pauli che agiscono su qubit diversi. Alcuni elementi di questa somma possono essere misurati tutti insieme; chiamiamo questo un gruppo di Pauli commutanti. A seconda del numero di termini non commutanti, potrebbero essere necessari molti di questi gruppi. Chiamiamo il numero di tali gruppi di stringhe di Pauli commutanti. Se è piccolo, questo approccio potrebbe funzionare bene. Se ha milioni di gruppi, non sarà utile.
I processi necessari per la stima del valore di aspettazione sono raccolti nella primitiva di Qiskit Runtime chiamata Estimator. Per saperne di più su Estimator, consulta il riferimento API nella documentazione IBM Quantum®. Si può usare Estimator direttamente, ma Estimator restituisce molto più del semplice autovalore energetico minimo. Per esempio, restituisce anche informazioni sull'errore standard dell'insieme. Quindi, nel contesto dei problemi di minimizzazione, si vede spesso Estimator all'interno di una funzione di costo. Per saperne di più sugli input e output di Estimator consulta questa guida nella documentazione IBM Quantum.
Registri il valore di aspettazione (o la funzione di costo) per l'insieme di parametri usato nel tuo stato, e poi aggiorni i parametri. Nel tempo, potresti usare i valori di aspettazione o di funzione di costo stimati per approssimare un gradiente della tua funzione di costo nel sottospazio degli stati campionati dal tuo ansatz. Esistono ottimizzatori classici sia basati sul gradiente che privi di gradiente. Entrambi soffrono di potenziali problemi di addestrabilità, come minimi locali multipli e ampie regioni dello spazio dei parametri con gradiente quasi zero, chiamate barren plateaus (altopiani desolati).

2.3 Fattori che determinano il costo computazionale
Il VQE non risolverà tutti i tuoi problemi più difficili di chimica quantistica. No. Ma non è questo il punto: non si tratta di essere migliori in tutti i calcoli. Abbiamo spostato ciò che determina il costo computazionale.

Siamo passati da un processo la cui complessità dipende solo dalla dimensione della matrice a uno che dipende dalla precisione richiesta e dal numero di operatori di Pauli non commutanti che compongono la matrice. Quest'ultimo aspetto non ha un analogo nel calcolo classico.
Sulla base di queste dipendenze, per matrici sparse, o matrici che coinvolgono poche stringhe di Pauli non commutanti, questo processo potrebbe essere utile. È il caso dei sistemi di spin interagenti, per esempio. Per le matrici dense, potrebbe essere meno utile. Sappiamo ad esempio che i sistemi chimici hanno spesso Hamiltoniani che coinvolgono centinaia, migliaia, persino milioni di stringhe di Pauli. Sono stati compiuti lavori interessanti per ridurre questo numero di termini. Ma i sistemi chimici potrebbero essere più adatti ad alcuni degli altri algoritmi che discuteremo in questo corso.
Verifica la tua comprensione
Considera un Hamiltoniano su quattro qubit che contiene i termini:
IIXX, IIXZ, IIZZ, IZXZ, IXXZ, ZZXZ, XZXZ, ZIXZ, ZZZZ, XXXX
Vuoi suddividere questi termini in gruppi in modo che tutti i termini in un gruppo possano essere misurati simultaneamente. Qual è il numero minimo di gruppi che puoi formare in modo che tutti i termini siano inclusi?
Risposta
Si può fare in 4 gruppi. Si noti che tali soluzioni tipicamente non sono uniche.
IIXX, XXXX, IIZZ, ZZZZ
IIXZ, IZXZ, ZIXZ, ZZXZ
IXXZ
XZXZ
Cosa ritieni che renda tipicamente difficile la chimica quantistica con il VQE: il numero di termini nell'Hamiltoniano, o trovare un buon ansatz?
Risposta
Si scopre che esistono ansätze altamente ottimizzati per contesti chimici. Il numero di termini nell'Hamiltoniano, e quindi il numero di misurazioni richieste, causa tipicamente più problemi.
3. Hamiltoniano di esempio
Mettiamo in pratica questo algoritmo utilizzando una piccola matrice hamiltoniana, in modo da poter vedere cosa accade in ogni passo. Utilizzeremo il framework dei pattern Qiskit:
-Passo 1: Mappa il problema su circuiti quantistici e operatori -Passo 2: Ottimizza per l'hardware di destinazione -Passo 3: Esegui sull'hardware di destinazione -Passo 4: Post-elabora i risultati
3.1 Passo 1: Mappa il problema su circuiti quantistici e operatori
Utilizzeremo quello definito sopra nel contesto della chimica. Iniziamo con alcuni import generali.
# General imports
import numpy as np
# SciPy minimizer routine
from scipy.optimize import minimize
# Plotting functions
import matplotlib.pyplot as plt
Anche qui, assumiamo che l'Hamiltoniano di interesse sia noto. Useremo un Hamiltoniano estremamente piccolo, perché altri metodi discussi in questo corso saranno più efficienti per risolvere problemi più grandi.
from qiskit.quantum_info import SparsePauliOp
import numpy as np
hamiltonian = SparsePauliOp.from_list(
[("YZ", 0.3980), ("ZI", -0.3980), ("ZZ", -0.0113), ("XX", 0.1810)]
)
A = np.array(hamiltonian)
eigenvalues, eigenvectors = np.linalg.eigh(A)
print("The ground state energy is ", min(eigenvalues))
The ground state energy is -0.702930394459531
Ci sono molte scelte di ansatz prefabbricate in Qiskit. Useremo efficient_su2.
# Pre-defined ansatz circuit and operator class for Hamiltonian
from qiskit.circuit.library import efficient_su2
# Note that it is more common to place initial 'h' gates outside the ansatz.
# Here we specifically wanted this layer structure.
ansatz = efficient_su2(
hamiltonian.num_qubits, su2_gates=["h", "rz", "y"], entanglement="circular", reps=1
)
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.decompose().draw("mpl", style="iqp")
This circuit has 4 parameters
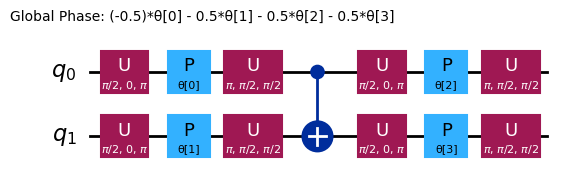
Ansätze diversi avranno strutture di entanglement diverse e gate di rotazione diversi. Quello mostrato qui usa gate CNOT per l'entanglement, e sia gate Y che gate RZ parametrizzati per le rotazioni. Nota le dimensioni di questo spazio dei parametri: significa che dobbiamo minimizzare la funzione di costo su 4 variabili (i parametri per i gate RZ). Questo può essere scalato, ma non indefinitamente. Eseguendo un problema simile su 4 qubit, usando i 3 reps predefiniti per efficient_su2 si ottengono 16 parametri variazionali.
3.2 Passo 2: Ottimizza per l'hardware di destinazione
L'ansatz è stato scritto usando gate familiari, ma il nostro circuito deve essere transpilato per fare uso dei gate base che possono essere implementati su ogni computer quantistico. Selezioniamo il backend meno occupato.
# runtime imports
from qiskit_ibm_runtime import QiskitRuntimeService, Session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# To run on hardware, select the backend with the fewest number of jobs in the queue
service = QiskitRuntimeService()
backend = service.least_busy(operational=True, simulator=False)
print(backend)
<IBMBackend('ibm_torino')>
Ora possiamo transpilare il nostro circuito per questo hardware e visualizzare il nostro ansatz transpilato.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")

Nota che i gate utilizzati sono cambiati, e i qubit nel nostro circuito astratto sono stati mappati su qubit con numerazione diversa sul computer quantistico. Dobbiamo mappare il nostro Hamiltoniano in modo identico affinché i risultati abbiano significato.
hamiltonian_isa = hamiltonian.apply_layout(layout=ansatz_isa.layout)
3.3 Passo 3: Esegui sull'hardware di destinazione
3.3.1 Registrazione dei valori
Definiamo qui una funzione di costo che accetta come argomenti le strutture costruite nei passi precedenti: i parametri, l'ansatz e l'Hamiltoniano. Usa anche l'Estimator, che non abbiamo ancora definito. Includiamo del codice per tracciare la storia della nostra funzione di costo, in modo da poter verificare il comportamento di convergenza.
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from Estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
È molto vantaggioso scegliere i valori iniziali dei parametri in base alla conoscenza del problema e alle caratteristiche dello stato target. Non faremo ipotesi su tale conoscenza e useremo valori iniziali casuali.
x0 = 2 * np.pi * np.random.random(num_params)
# This required 13 min, 20 s QPU time on an Eagle processor, 28 min total time.
with Session(backend=backend) as session:
estimator = Estimator(mode=session)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 50},
)
Iters. done: 1 [Current cost: 0.010575798722044727]
Iters. done: 2 [Current cost: 0.004040015974440895]
Iters. done: 3 [Current cost: 0.0020213258785942503]
Iters. done: 4 [Current cost: 0.18723082446726014]
Iters. done: 5 [Current cost: -0.2746792152068885]
Iters. done: 6 [Current cost: -0.3094547651648519]
Iters. done: 7 [Current cost: -0.05281985428356641]
Iters. done: 8 [Current cost: 0.00808560303514377]
Iters. done: 9 [Current cost: -0.0014821685303514388]
Iters. done: 10 [Current cost: -0.004759824281150161]
Iters. done: 11 [Current cost: 0.09942328705995292]
Iters. done: 12 [Current cost: 0.01092366214057508]
Iters. done: 13 [Current cost: 0.05017497496069776]
Iters. done: 14 [Current cost: 0.13028868414310696]
Iters. done: 15 [Current cost: 0.013747803514376994]
Iters. done: 16 [Current cost: 0.2583072432944498]
Iters. done: 17 [Current cost: -0.14422125655131562]
Iters. done: 18 [Current cost: -0.0004950150347678081]
Iters. done: 19 [Current cost: 0.00681082268370607]
Iters. done: 20 [Current cost: -0.0023377795527156544]
Iters. done: 21 [Current cost: 0.6027665591169237]
Iters. done: 22 [Current cost: 0.00596641373801917]
Iters. done: 23 [Current cost: -0.008318769968051117]
Iters. done: 24 [Current cost: -0.00026683306709265246]
Iters. done: 25 [Current cost: -0.007648222843450479]
Iters. done: 26 [Current cost: 0.004121086261980831]
Iters. done: 27 [Current cost: -0.004075019968051117]
Iters. done: 28 [Current cost: -0.004419369009584665]
Iters. done: 29 [Current cost: 0.213185460054037]
Iters. done: 30 [Current cost: -0.06505919572162797]
Iters. done: 31 [Current cost: -0.5334241316590271]
Iters. done: 32 [Current cost: 0.00218370607028754]
Iters. done: 33 [Current cost: 0.09579352143666908]
Iters. done: 34 [Current cost: -0.009274800319488819]
Iters. done: 35 [Current cost: -0.44395141360688106]
Iters. done: 36 [Current cost: 0.011747104632587858]
Iters. done: 37 [Current cost: -0.003344149361022364]
Iters. done: 38 [Current cost: 0.19138183916486304]
Iters. done: 39 [Current cost: 0.013513931813145209]
Possiamo esaminare gli output grezzi.
res
message: Return from COBYLA because the trust region radius reaches its lower bound.
success: True
status: 0
fun: -0.5334241316590271
x: [ 1.024e+00 6.459e+00 3.625e+00 4.007e+00]
nfev: 39
maxcv: 0.0
3.4 Passo 4: Post-elabora i risultati
Se la procedura termina correttamente, i valori nel nostro dizionario dovrebbero essere uguali al vettore soluzione e al numero totale di valutazioni della funzione, rispettivamente. È facile verificarlo:
cost_history_dict
{'prev_vector': array([1.02397956, 6.45886604, 3.62479262, 4.00744128]),
'iters': 39,
'cost_history': [np.float64(0.010575798722044727),
np.float64(0.004040015974440895),
np.float64(0.0020213258785942503),
np.float64(0.18723082446726014),
np.float64(-0.2746792152068885),
np.float64(-0.3094547651648519),
np.float64(-0.05281985428356641),
np.float64(0.00808560303514377),
np.float64(-0.0014821685303514388),
np.float64(-0.004759824281150161),
np.float64(0.09942328705995292),
np.float64(0.01092366214057508),
np.float64(0.05017497496069776),
np.float64(0.13028868414310696),
np.float64(0.013747803514376994),
np.float64(0.2583072432944498),
np.float64(-0.14422125655131562),
np.float64(-0.0004950150347678081),
np.float64(0.00681082268370607),
np.float64(-0.0023377795527156544),
np.float64(0.6027665591169237),
np.float64(0.00596641373801917),
np.float64(-0.008318769968051117),
np.float64(-0.00026683306709265246),
np.float64(-0.007648222843450479),
np.float64(0.004121086261980831),
np.float64(-0.004075019968051117),
np.float64(-0.004419369009584665),
np.float64(0.213185460054037),
np.float64(-0.06505919572162797),
np.float64(-0.5334241316590271),
np.float64(0.00218370607028754),
np.float64(0.09579352143666908),
np.float64(-0.009274800319488819),
np.float64(-0.44395141360688106),
np.float64(0.011747104632587858),
np.float64(-0.003344149361022364),
np.float64(0.19138183916486304),
np.float64(0.013513931813145209)]}
fig, ax = plt.subplots()
x = np.linspace(0, 10, 50)
# Define the constant function
constant = -0.7029
y_constant = np.full_like(x, constant)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

IBM Quantum offre altre risorse di aggiornamento professionale relative al VQE. Se sei pronto a mettere in pratica il VQE, consulta il nostro tutorial: Stima dell'energia del ground state della catena di Heisenberg con VQE. Se vuoi maggiori informazioni sulla creazione di Hamiltoniani molecolari, consulta questa lezione nel nostro corso su Chimica quantistica con VQE. Se sei interessato a una comprensione più approfondita del funzionamento degli algoritmi variazionali come il VQE, ti consigliamo il corso Variational Algorithm Design.
Verifica la tua comprensione
In questa sezione abbiamo calcolato un'energia del ground state da un Hamiltoniano. Se volessimo applicarlo, ad esempio, alla determinazione della geometria di una molecola, come estenderemmo questo approccio?
Risposta
Dovremmo introdurre variabili per la spaziatura interatomica e gli angoli tra i legami. Dovremmo variare questi parametri. Per ogni variazione di questi, produrremmo un nuovo Hamiltoniano (poiché gli operatori che descrivono l'energia dipendono certamente dalla geometria). Per ogni tale Hamiltoniano prodotto e mappato sui qubit, dovremmo effettuare un'ottimizzazione come quella mostrata sopra. Di tutti quei numerosi problemi di ottimizzazione convergenti, la geometria che ha prodotto l'energia più bassa sarebbe quella adottata dalla natura. Questo è molto più complesso di quanto mostrato sopra. Tale calcolo viene eseguito per la molecola più semplice, , qui.
4. Relazione del VQE con altri metodi
In questa sezione esamineremo i vantaggi e gli svantaggi dell'approccio VQE originale e indicheremo le sue relazioni con altri algoritmi più recenti.
4.1 I punti di forza e di debolezza del VQE
Alcuni punti di forza sono già stati evidenziati. Includono:
- Adattabilità all'hardware moderno: Alcuni algoritmi quantistici richiedono tassi di errore molto più bassi, avvicinandosi a una tolleranza agli errori su larga scala. Il VQE no; può essere implementato su computer quantistici attuali.
- Circuiti superficiali: Il VQE impiega spesso circuiti quantistici relativamente superficiali. Questo rende il VQE meno suscettibile agli errori accumulati dei gate e lo rende adatto a molte tecniche di mitigazione degli errori. Naturalmente, i circuiti non sono sempre superficiali; questo dipende dall'ansatz utilizzato.
- Versatilità: Il VQE può (in linea di principio) essere applicato a qualsiasi problema che possa essere formulato come problema di autovalori/autovettori. Esistono molte avvertenze che rendono il VQE impraticabile o svantaggioso per alcuni problemi. Alcune di queste sono riepilogate di seguito.
Alcuni punti di debolezza del VQE e problemi per cui è impraticabile sono stati descritti sopra. Questi includono:
- Natura euristica: Il VQE non garantisce la convergenza alla corretta energia del ground state, poiché le sue prestazioni dipendono dalla scelta dell'ansatz e dei metodi di ottimizzazione[1-2]. Se viene scelto un ansatz inadeguato privo dell'entanglement necessario per il ground state desiderato, nessun ottimizzatore classico può raggiungere quel ground state.
- Parametri potenzialmente numerosi: Un ansatz molto espressivo può avere così tanti parametri che le iterazioni di minimizzazione richiedono molto tempo.
- Elevato overhead di misura: Nel VQE, viene utilizzato Estimator per stimare il valore atteso di ciascun termine dell'Hamiltoniano. La maggior parte degli Hamiltoniani di interesse avrà termini che non possono essere stimati simultaneamente. Questo può rendere il VQE molto oneroso in termini di risorse per sistemi grandi con Hamiltoniani complicati[1].
- Effetti del rumore: Quando l'ottimizzatore classico cerca un minimo, calcoli rumorosi possono confonderlo e allontanarlo dal vero minimo o ritardarne la convergenza. Una possibile soluzione è sfruttare le tecniche all'avanguardia di mitigazione e soppressione degli errori[2-3] di IBM.
- Pianure desolate (barren plateaus): Queste regioni di gradienti che si annullano[2-3] esistono anche in assenza di rumore, ma il rumore le rende più problematiche poiché la variazione dei valori attesi dovuta al rumore potrebbe essere maggiore della variazione derivante dall'aggiornamento dei parametri in queste regioni deserte.
4.2 Relazione con altri approcci
Adapt-VQE
L'algoritmo ADAPT-VQE (Adaptive Derivative-Assembled Pseudo-Trotter Variational Quantum Eigensolver) è un miglioramento dell'algoritmo VQE originale, progettato per migliorare l'efficienza, l'accuratezza e la scalabilità delle simulazioni quantistiche, in particolare nella chimica quantistica.
L'algoritmo VQE originale descritto in questa lezione usa un ansatz predefinito e fisso per approssimare il ground state del sistema. Nel nostro caso, abbiamo usato efficient_su2, con una singola ripetizione, usando gate di rotazione Y e RZ. Sebbene i parametri nei gate RZ siano cambiati, la struttura di questo ansatz e i gate utilizzati non sono cambiati.
ADAPT-VQE affronta i limiti del VQE attraverso la costruzione adattiva dell'ansatz. Invece di partire da un ansatz fisso, ADAPT-VQE costruisce dinamicamente l'ansatz in modo iterativo. Ad ogni passo, seleziona l'operatore da un pool predefinito (come gli operatori di eccitazione fermionica) che ha il gradiente più grande rispetto all'energia. Ciò garantisce che vengano aggiunti solo gli operatori più impattanti, portando a un ansatz compatto ed efficiente[4-6]. Questo approccio può avere diversi effetti benefici:
- Riduzione della profondità del circuito: Aumentando l'ansatz in modo incrementale e concentrandosi solo sugli operatori necessari, ADAPT-VQE minimizza le operazioni di gate rispetto agli approcci VQE tradizionali[5,7].
- Maggiore accuratezza: La natura adattiva consente a ADAPT-VQE di recuperare più energia di correlazione ad ogni passo, rendendolo particolarmente efficace per sistemi fortemente correlati in cui il VQE tradizionale fatica[8,9].
- Scalabilità e robustezza al rumore: L'ansatz compatto riduce l'accumulo di errori dei gate, riduce l'overhead computazionale e limita il numero di parametri variazionali che devono essere minimizzati.
ADAPT-VQE non è ancora perfetto. In alcuni casi può rimanere intrappolato o rallentato da minimi locali, e può soffrire di sovra-parametrizzazione. Può anche essere piuttosto intensivo in termini di risorse, poiché richiede il calcolo dei gradienti e l'ottimizzazione dei parametri con molte strutture di gate.
Quantum phase estimation (QPE)
QPE ha uno scopo simile al VQE, ma un'implementazione molto diversa. QPE richiede computer quantistici fault-tolerant a causa dei suoi circuiti quantistici generalmente profondi e dell'alto livello di coerenza che richiede. Una volta che QPE potrà essere implementato, sarebbe più preciso del VQE. Un modo per descrivere la differenza è attraverso la precisione in funzione della profondità del circuito. QPE raggiunge una precisione con profondità di circuito scalanti come [10]. Il VQE richiede campioni per raggiungere la stessa precisione[10,11].
Krylov, SQD, QSCI e altri in questo corso
Il VQE ha contribuito a stabilire algoritmi quantistici che dipendono ancora dai computer classici, non solo per far funzionare il computer quantistico, ma per parti sostanziali dell'algoritmo. Diversi di tali algoritmi sono al centro della parte restante di questo corso. Qui forniamo una spiegazione sommaria di alcuni, semplicemente per confrontarli e contrastarli con il VQE. Verranno spiegati in modo molto più dettagliato nelle lezioni successive.
Krylov quantum diagonalization (KQD)
I metodi del sottospazio di Krylov sono modi di proiettare una matrice su un sottospazio per ridurne la dimensione e renderla più gestibile, mantenendo le caratteristiche più importanti. Un trucco in questo metodo è generare un sottospazio che conservi queste caratteristiche; si scopre che la generazione di questo sottospazio è strettamente correlata a un metodo ben consolidato su computer quantistici chiamato Trotterizzazione.
Esistono alcune varianti dei metodi Krylov quantistici, ma in generale l'approccio è:
- Utilizzare il computer quantistico per generare un sottospazio (il sottospazio di Krylov) attraverso la Trotterizzazione
- Proiettare la matrice di interesse su quel sottospazio di Krylov
- Diagonalizzare il nuovo Hamiltoniano proiettato usando un computer classico
Sampling-based quantum diagonalization (SQD)
La diagonalizzazione quantistica basata sul campionamento (SQD) è correlata al metodo di Krylov in quanto tenta anche di ridurre la dimensione di una matrice da diagonalizzare preservando le caratteristiche chiave. SQD lo fa nel modo seguente:
- Inizia con un'ipotesi iniziale per il tuo ground state e prepara il sistema in quel ground state.
- Usa Sampler per campionare le bitstring che compongono questo stato.
- Usa la collezione di stati della base computazionale del sampler come sottospazio su cui proiettare la matrice di interesse.
- Diagonalizza la matrice proiettata più piccola usando un computer classico.
Questo è correlato al VQE in quanto sfrutta il calcolo classico e quantistico per componenti sostanziali dell'algoritmo. Entrambi condividono anche il requisito di preparare una buona ipotesi iniziale o ansatz. Ma la distribuzione del lavoro tra i computer classico e quantistico in SQD è più simile a quella del metodo di Krylov.
In effetti, il metodo di Krylov e SQD sono stati recentemente combinati nel metodo di diagonalizzazione quantistica di Krylov basato sul campionamento (SKQD) [12].
Quantum subspace configuration interaction
La Quantum Selected Configuration Interaction (QSCI)[13] è un algoritmo che produce uno stato fondamentale approssimato di un Hamiltoniano campionando una funzione d'onda di prova per identificare gli stati della base computazionale significativi al fine di generare un sottospazio per una diagonalizzazione classica. Sia SQD che QSCI utilizzano un computer quantistico per costruire un sottospazio ridotto. Il punto di forza aggiuntivo di QSCI risiede nella sua preparazione dello stato, specialmente nel contesto dei problemi di chimica. Sfrutta varie strategie come l'uso di stati evoluti nel tempo [14] e un insieme di ansätze ispirati alla chimica. Concentrandosi su una preparazione efficiente degli stati, QSCI riduce i costi computazionali quantistici per gli Hamiltoniani chimici mantenendo un'alta fedeltà e sfruttando la robustezza al rumore derivante dalle tecniche di campionamento degli stati quantistici [15]. QSCI fornisce anche una tecnica di costruzione adattiva che offre più ansätze per un risultato migliore.
Il workflow predefinito di QSCI per i problemi di chimica è il seguente:
- Costruisci l'Hamiltoniano molecolare usando il software di tua scelta (come SciPy).
- Prepara un algoritmo QSCI selezionando uno stato iniziale appropriato e un ansatz ispirato alla chimica con un insieme di parametri pre-selezionati.
- Campiona gli stati della base significativi e diagonalizza l'Hamiltoniano usando un computer classico per ottenere l'energia del ground state.
- Spesso si usa il configuration recovery [16] e la symmetry postselection [15] come tecnica di post-elaborazione.
- Opzionalmente, il workflow del QSCI adattivo ha un ciclo di ottimizzazione aggiuntivo dal passo 2 al passo 3, usando più ansätze con stati iniziali casuali.
Verifica la tua comprensione
Cosa ha in comune il VQE con tutti gli altri metodi elencati sopra (eccetto QPE che non è descritto in dettaglio)
Risposta
Tutti implicano uno stato di prova o una funzione d'onda di qualche tipo. Tutti funzionano meglio quando l'ipotesi iniziale per questo stato di prova è eccellente.
Un'altra risposta corretta è che sono tutti più facili da implementare quando l'Hamiltoniano è facile da misurare (può essere suddiviso in un numero relativamente piccolo di gruppi di operatori di Pauli che commutano).
Cosa ha il VQE che nessuno degli altri metodi elencati sopra ha?
Risposta
Gli ottimizzatori classici. Nessuno degli altri usa algoritmi di ottimizzazione classici per selezionare i parametri variazionali.
Riferimenti
[2] https://en.wikipedia.org/wiki/Variational_quantum_eigensolver
[3] https://journals.aps.org/prapplied/abstract/10.1103/PhysRevApplied.19.024047
[4] https://arxiv.org/abs/2111.05176
[6] https://inquanto.quantinuum.com/tutorials/InQ_tut_fe4n2_2.html
[7] https://www.nature.com/articles/s41467-019-10988-2
[8] https://arxiv.org/abs/2210.15438
[9] https://journals.aps.org/prresearch/abstract/10.1103/PhysRevResearch.6.013254
[10] https://arxiv.org/html/2403.09624v1
[11] https://www.nature.com/articles/s42005-023-01312-y
[13] https://arxiv.org/abs/1802.00171
[14] https://arxiv.org/abs/2103.08505
[15] https://arxiv.org/html/2501.09702v1
[16] https://quri-sdk.qunasys.com/docs/examples/quri-algo-vm/qsci/