Mappatura
Guarda il video sul mapping di Olivia Lanes, oppure aprilo in una finestra separata su YouTube.
Introduzione
In questa lezione ci concentreremo sul primo passo — spesso il più impegnativo — nella definizione di un programma quantistico: il mapping di un problema da eseguire su un computer quantistico. Questo passaggio descrive come si parte da un problema computazionale e lo si traduce in qualcosa che può essere risolto su un computer quantistico.
Nelle Lezioni 2 e 3 di questo corso abbiamo menzionato che la fase di mapping è la prima delle quattro fasi totali del framework Qiskit patterns. Da quelle lezioni potresti ricordare che l'obiettivo del mapping è tradurre o riscrivere un problema computazionale in una funzione di costo o valore di aspettazione che possiamo valutare usando un computer quantistico.
Nella Lezione 3 abbiamo discusso un esempio concreto con il Max-Cut, un problema computazionalmente difficile ma molto comune nell'ottimizzazione combinatoria. In quell'esempio abbiamo percorso diversi passi per tradurre il problema iniziale su grafi in uno risolvibile su un computer quantistico. Abbiamo trasformato il problema di trovare il numero massimo di tagli nel grafo in una funzione di costo, riscritto quella funzione di costo come Hamiltoniano, e poi preparato uno stato quantistico di prova il cui stato fondamentale corrispondeva al taglio massimo. Infine, abbiamo costruito un circuito quantistico che rappresenta lo stato quantistico di prova di interesse, aggiungendo poi i gate specifici per permettere allo stato di evolvere nel tempo. Questa sequenza di passi faceva parte interamente del mapping. Sebbene i passi esatti fossero specifici al problema Max-Cut, la stessa procedura generale può essere applicata a molte altre applicazioni, come la chimica quantistica e le simulazioni quantistiche.
Il mapping può essere difficile. Non esiste una strategia unica valida per ogni singolo problema, il che può risultare scoraggiante. In questa lezione vedremo alcune considerazioni generali sul mapping, per poi addentrarci in alcuni problemi esemplificativi che illustrano i vari modi di mappare un problema su un computer quantistico.
Considerazioni generali
Sebbene la strategia esatta per mappare un problema su un computer quantistico dipenda dal problema in questione, in genere si raggiungono un paio di obiettivi principali.
Per prima cosa, potrebbe essere necessario semplificare il problema per renderlo fattibile. Questo non è esclusivo del mondo quantistico — tutte le discipline scientifiche usano modelli semplificati per studiare i fenomeni di interesse, ignorando i dettagli irrilevanti. In fisica c'è una famosa espressione che porta questo principio all'estremo: "assumere una mucca sferica." Spesso è troppo difficile descrivere un sistema esattamente come appare, ma possiamo invece fare semplificazioni ragionevoli che possono comunque portare a soluzioni utili. Alcuni esempi di come potremmo farlo nel quantum computing sono: limitare la dimensione o la profondità del circuito scegliendo un ansatz hardware-efficiente, troncare evoluzioni temporali complesse, o trascurare i termini Hamiltoniani che contribuiscono poco all'energia finale di uno stato quantistico.
In secondo luogo, il mapping implica scrivere il problema in modo che un computer quantistico possa comprenderlo. Spesso ciò comporta porsi queste tre domande:
- Cosa rappresenteranno i nostri qubit nel nostro modello?
- Il nostro problema è continuo? Poiché i computer quantistici sono digitali, se il problema è continuo dovremo trovare un modo per discretizzarlo.
- Infine, la topologia del problema si allinea con la topologia dell'hardware? In caso contrario, potremmo dover implementare alcuni accorgimenti per farlo funzionare.
Affrontiamo la prima domanda, che è al cuore di molte delle difficoltà legate alla comprensione del mapping: cosa può rappresentare un qubit?
I qubit possono essere usati per rappresentare molte cose. La prima, e forse la più semplice, è un nodo su un grafo. I grafi sono usati per mostrare la connettività in molti tipi diversi di problemi matematici, e i nodi sono un elemento fondamentale che rappresenta un punto o un'entità all'interno di una rete. A seconda di cosa rappresenta l'intera rete, potrebbe trattarsi di una città, una persona, o un ferromagnete in un reticolo.
I qubit possono essere usati anche per rappresentare bosoni e fermioni, anche se avverto che un singolo qubit non equivale esattamente a un bosone o a un fermione — è un po' più complicato di così, come discuteremo più avanti nella lezione.
Ora arriviamo ad esempi un po' più complicati. Per questi modelli non ha più senso parlare di singoli qubit; invece abbiamo bisogno di gruppi di qubit per formare qualcosa di fisico. Per esempio, un gruppo di qubit, qui rappresentati su una topologia heavy-hexagonal, può essere usato per rappresentare le posizioni geometriche degli aminoacidi; catene polimeriche. Un altro esempio è la simulazione dello scattering adronico in modelli di fisica delle alte energie, che può essere realizzata simulando l'evoluzione temporale di un pacchetto d'onda adronico. In questo caso, un registro di qubit può essere usato per codificare diversi stati di un campo quantistico: lo stato di vuoto di quel campo, o un pacchetto d'onda che si propaga su quel vuoto.
Ma a questo punto abbiamo parlato in modo abbastanza astratto della sfida che ci attende. Analizziamo questi esempi nel dettaglio.
Esempi di mapping
Max-Cut
Cominciamo con il primo esempio. Uno dei problemi di mapping più semplici è quello che abbiamo già affrontato in una certa misura: l'esempio del Max-Cut. In quel problema il mapping era piuttosto semplice per noi perché un qubit equivaleva a un nodo del nostro grafo.
Ricorda che, per mappare il problema Max-Cut, abbiamo espresso la funzione di costo come un Hamiltoniano usando la formulazione QUBO. Una funzione di costo Hamiltoniana è una funzione che codifica la soluzione ottimale del problema nello stato fondamentale dell'Hamiltoniano. Per costruire l'Hamiltoniano di costo, abbiamo usato la classe SparsePauliOp in Qiskit per specificare la connettività del nostro grafo, e la fase di mapping agli operatori quantistici era completata. E il circuito quantistico era semplicemente l'ansatz QAOA. Per un ripasso, consulta la lezione QAOA su scala utility, dove percorriamo tutto questo in molto maggior dettaglio.
In quella lezione, anche nell'esempio su scala utility a 100 qubit, la connettività del grafo corrispondeva già alla topologia del nostro hardware superconduttore. Quindi non abbiamo dovuto preoccuparci di come gestire topologie diverse. Ma non è sempre così. Se avessimo un grafo più complicato o densamente connesso rispetto agli esempi evidenziati finora, dovremmo implementare una serie di gate SWAP per modificare la connettività effettiva dell'hardware. Questo viene gestito al secondo passo di Qiskit patterns, la transpilazione, ma dovrebbe essere tenuto a mente già nella fase di mapping.
Ripiegamento delle proteine
Esploriamo ora un esempio modellato nel paper intitolato "Resource-efficient quantum algorithm for protein folding," scritto da IBM® e collaboratori dell'University of New South Wales.
Un po' di background di biochimica: le proteine sono macromolecole composte da lunghe catene di aminoacidi. Queste catene si ripiegano in strutture complesse che svolgono un'ampia varietà di funzioni biologiche. Determinare la struttura tridimensionale di una proteina e comprendere le relazioni tra struttura e funzione sono tra i problemi più impegnativi della biochimica moderna. Le proteine si ripiegano in strutture funzionali a causa delle interazioni tra aminoacidi. Man mano che la struttura si torce e si piega, aminoacidi lontani tra loro lungo la catena possono finire uno accanto all'altro e interagire fortemente.
Per modellare questo su un computer quantistico, abbiamo bisogno di un Hamiltoniano che descriva tutte queste interazioni tra aminoacidi. Poi, possiamo prevedere la struttura finale trovando lo stato che minimizzerà l'energia del nostro Hamiltoniano. Qui ci concentreremo su come le catene di aminoacidi possono essere modellate su un computer quantistico e su come possiamo ottenere le distanze inter-aminoacidiche per il calcolo delle energie di interazione. Con questo avremo raccolto tutti i contributi necessari all'Hamiltoniano che è necessario simulare su un computer quantistico.
Nelle proteine reali, gli aminoacidi possono occupare una serie continua di posizioni possibili. Tuttavia, useremo una semplificazione e li vincolaremo usando un modello a reticolo, dove ogni aminoacido occupa un punto su una griglia. Qui, gli autori hanno usato un reticolo tetraedrico. Nota rapida: qui stiamo codificando la direzione degli archi, non i nodi stessi come nel problema Max-Cut. Ogni qubit rappresenta un possibile percorso a singolo passo lungo la griglia tetraedrica. Nota che i siti adiacenti sono stati etichettati come A o B a causa dei loro diversi orientamenti nel reticolo.
La catena proteica è rappresentata come una serie di svolte o direzioni su questo reticolo. Ogni svolta tra aminoacidi può essere in una delle quattro direzioni, corrispondenti agli archi del tetraedro. Queste quattro possibili svolte sono codificate usando quattro qubit negli stati 0001, 0010, 0100, o 1000.
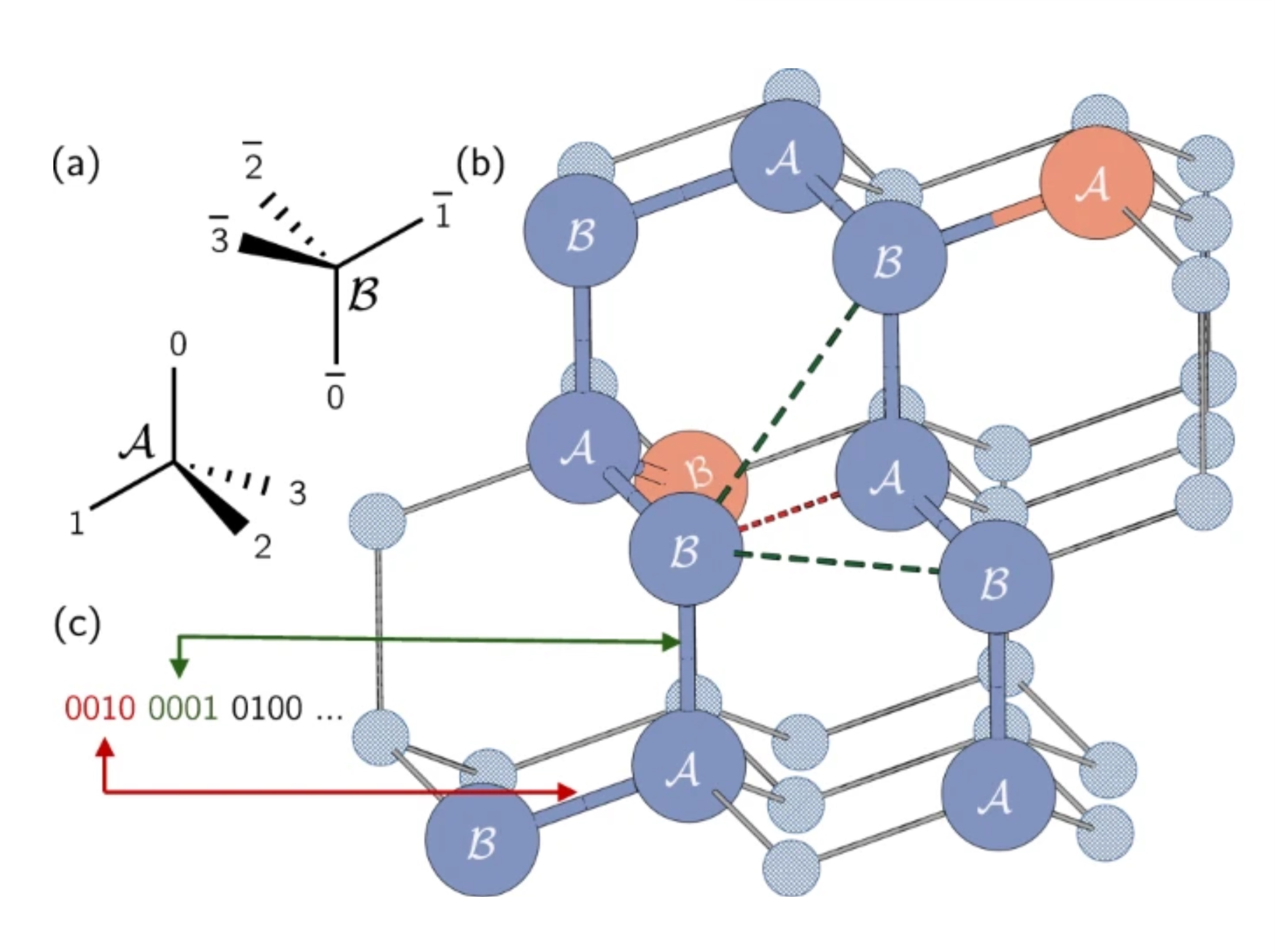
Guardiamo un esempio nella figura sopra. Posizioniamo il nostro primo aminoacido sul punto etichettato "B" cerchiato in rosso nel nostro reticolo tetraedrico. La direzione dal primo aminoacido al secondo è arbitraria perché il sistema può sempre essere ruotato per far puntare quell'arco in qualsiasi direzione vogliamo. Quindi, possiamo posizionare il nostro secondo aminoacido sul punto sotto il primo etichettato "A". Non è altrettanto facile da vedere, ma il percorso dal secondo al terzo è anch'esso arbitrario. Tutte e tre le scelte risulterebbero nell'avere due archi con un angolo di circa 109,5 gradi tra loro. Scegliere questo secondo arco determina semplicemente l'orientamento della nostra proteina nello spazio. Quindi, senza perdita di generalità, possiamo scegliere che le prime due svolte siano semplicemente 0001 e 0010.
Con queste semplificazioni, la configurazione della catena di aminoacidi è data da questa espressione:
Finora abbiamo mappato gli archi del tetraedro sui qubit, e il nostro circuito quantistico sarà un ansatz. Ora dobbiamo considerare come codificare l'energia del problema in un Hamiltoniano, in modo che il suo stato fondamentale ci dia il pattern di ripiegamento ottimale.
Per qualsiasi configurazione data, ci sarà un'energia associata dovuta alle interazioni tra gli aminoacidi. Queste interazioni sono più forti quando i due aminoacidi sono vicini tra loro. Ovviamente, gli aminoacidi adiacenti nella backbone della catena interagiranno sempre tra loro. Ma poiché la proteina può torcersi e piegarsi, anche altre coppie di aminoacidi possono interagire. Gli aminoacidi 10 e 20, ad esempio, potrebbero finire su siti adiacenti dopo che la proteina si è ripiegata. Quindi, abbiamo bisogno di una formula per descrivere la distanza tra gli aminoacidi e usando le informazioni codificate sui qubit di configurazione. In questo modo possiamo usare la loro distanza di separazione per determinare quanto fortemente interagiscono.
Per prima cosa, introduciamo una funzione che indica se un arco viene usato o meno per la svolta all'aminoacido . Qui può assumere uno qualsiasi delle quattro possibili direzioni. Sulla base della configurazione con cui abbiamo iniziato sopra, possiamo scrivere queste funzioni:
Poi possiamo definire una differenza nel numero di svolte etichettate sui reticoli A e B dall'indice all'indice come :
Perché fare questo? Ebbene, si scopre che una svolta di dal sito di reticolo A al B viene esattamente annullata da una svolta di dal sito di reticolo B ad A. Quindi, per conoscere la distanza dell'aminoacido sul sito da quello sul sito , dobbiamo solo trovare la differenza tra i passi effettuati lungo gli archi dai siti A e dai siti B. Poiché i siti A e B si alternano necessariamente lungo la backbone della proteina, questo è catturato nel . Lo stesso argomento si applica a tutti e quattro i tipi di archi. Quindi, la distanza totale tra gli aminoacidi in passi sul reticolo tetraedrico può essere calcolata con questa espressione:
Ma come otteniamo l'Hamiltoniano da questa lunga equazione per la distanza totale tra gli aminoacidi? Prima di tutto, possiamo convertire dalla distanza in passi sul reticolo allo spazio euclideo con un po' di geometria semplice:
Queste distanze verranno poi usate nel calcolo dell'energia della configurazione proteica. A seconda dei nostri obiettivi, potremmo stabilire una distanza limite al di sotto della quale consideriamo la coppia in interazione, oppure potremmo fare qualcosa di più complicato.
Potrebbe non essere ovvio, ma in realtà con questo abbiamo finito la fase di mapping. Gli stati dei qubit indicano la "svolta" della proteina in ogni sito del reticolo, e l'insieme delle svolte determina la distanza tra qualsiasi coppia di aminoacidi. Coppie di varie specie di aminoacidi hanno interazioni diverse, alcune attrattive, alcune repulsive. Se stai usando la configurazione e le distanze semplicemente per determinare se le interazioni note tra aminoacidi sono "attive" o "disattive", le forze di queste sono già state ricavate e possono semplicemente essere cercate in una tabella come questa:
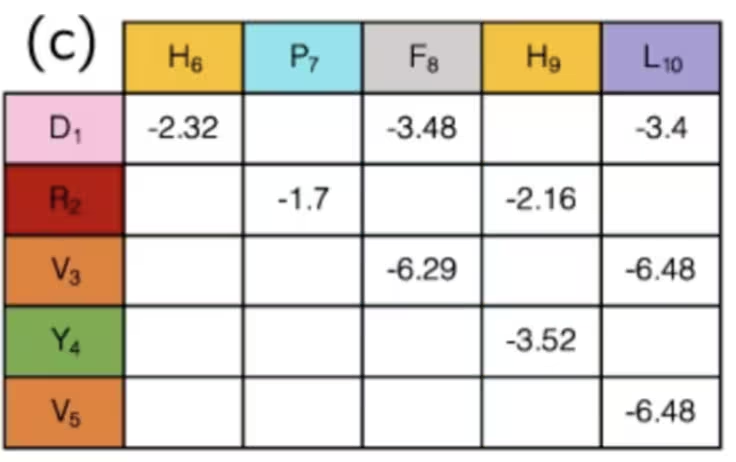
In sintesi, in questo esempio, i qubit vengono usati per contrassegnare i passi in un percorso lungo un reticolo che, insieme, formano una catena di aminoacidi. Simulando come si piegano e si ripiegano, possiamo sperare di trovare risultati migliori nella ricerca medica. Abbiamo saltato il calcolo di alcuni termini di questo Hamiltoniano perché erano iper-specifici a questo problema, mentre la definizione di direzioni su un reticolo può essere applicata più in generale. Una volta ottenuto un Hamiltoniano generale, si vorrà sempre tradurlo in operatori di Pauli, che sono nativi del computer quantistico. Di questo parleremo di seguito.
Trasformazione di Jordan-Wigner
Vediamo ora come tradurre un sistema di particelle subatomiche in operatori di Pauli.
Le particelle subatomiche si dividono in due categorie: bosoni e fermioni. I bosoni, come i fotoni o il bosone di Higgs, obbediscono a un certo insieme di regole statistiche. I fermioni, come gli elettroni o i neutrini, ne seguono un altro. La differenza fondamentale tra loro è che i bosoni possono occupare lo stesso stato — non c'è limite al numero di bosoni che possono trovarsi nello stato fondamentale o in qualsiasi stato eccitato. I fermioni, invece, sono "egoisti" — richiedono che ogni singola particella abbia il proprio stato quantistico.
I bosoni hanno anche spin intero mentre i fermioni hanno spin semi-intero, come l'elettrone con spin-1/2, e particelle più esotiche con spin-3/2. Per descrivere un sistema di particelle, abbiamo bisogno di una descrizione della loro energia. Concentriamoci sui fermioni. Possiamo partire da un Hamiltoniano scritto in termini di quelli che chiamiamo operatori c. Questi sono fondamentalmente oggetti matematici che corrispondono alla creazione o all'annichilazione di un fermione in uno stato del sistema. Sono spesso scritti come e , dove è l'operatore che crea un fermione nello stato e è l'operatore che distrugge un fermione nello stato .
Ricorda però che i computer quantistici di solito operano in una base di qubit con regole specifiche per rappresentare i sistemi fermionici; non lavorano intrinsecamente nel linguaggio degli operatori fermionici. Per colmare questo divario, dobbiamo mappare questa notazione fermionica sugli operatori di Pauli, che corrispondono naturalmente ai gate quantistici.
Esistono diverse trasformazioni di questo tipo per i fermioni. Una scelta comune è la trasformazione di Jordan-Wigner. Le mappature di Bravyi-Kitaev e di parità sono codifiche fermioniche più recenti. Gli operatori bosonici possono essere trasformati usando la trasformazione di Holstein-Primakoff o mappando direttamente gli stati di Fock a una sotto-base dei modi bosonici disponibili, tra le altre opzioni. Sono attivamente in corso ricerche anche su altre codifiche. Per ora ci concentreremo solo sulla trasformazione di Jordan-Wigner.
La trasformazione di Jordan-Wigner implica la mappatura di un singolo fermione su molti qubit. Perché non possiamo semplicemente assegnare un qubit a ciascun elettrone? Questo ha a che fare con la distinguibilità di fermioni identici. I qubit sono distinguibili mentre gli elettroni non lo sono. Per esempio, possiamo facilmente etichettare e identificare i singoli qubit su qualsiasi dispositivo. Ma l'indistinguibilità degli elettroni significa che non possiamo etichettarli affatto. Pertanto, dobbiamo effettivamente etichettare gli operatori in base agli orbitali occupati, come 1s, 2p, 2p, ecc., invece degli elettroni specifici. Quindi, ogni qubit svolge approssimativamente il ruolo di un orbitale nella molecola che è occupato o non occupato. Ma come farlo è un po' più complicato. La mappatura di Jordan-Wigner tiene traccia dell'anti-simmetria e garantisce le corrette statistiche dell'intero sistema fermionico. La mappatura di Jordan-Wigner esprime gli operatori fermionici in termini di operatori di Pauli usando queste relazioni:
La mappatura di Jordan-Wigner è concettualmente semplice grazie alla corrispondenza uno a uno tra orbitali e qubit. Esistono altre mappature che raggiungono un obiettivo simile, tra cui la mappatura di parità. Il calcolo della parità di uno stato richiede la considerazione di più qubit. Nella mappatura di parità (e in alcune altre) l'interpretazione di un qubit corrispondente a un orbitale non è valida. Percorriamo ora un semplice esempio. Supponiamo di voler calcolare l'interazione a singolo qubit . Iniziamo inserendo le nostre definizioni per gli operatori di creazione e annichilazione.
Il commutatore . Quindi, l'espressione finale diventa:
Abbiamo così riscritto con successo un'espressione fermionica in termini di operatori di Pauli che il nostro computer quantistico sarà in grado di comprendere.
Vediamo rapidamente come implementare la mappatura di Jordan-Wigner in Qiskit. È importante capire come funzionano questo tipo di trasformazioni, ma sarebbe poco pratico calcolarle manualmente ogni volta — specialmente per sistemi di grandi dimensioni. Fortunatamente, Qiskit semplifica tutto questo con la funzione SparsePauliOp.
Ad alto livello, i passi sono:
- Usa la funzione
from_listdi SparsePauliOp per creare un operatore identità corrispondente alla dimensione dello spazio dei parametri da mappare. - Seguendo la definizione degli operatori di creazione e distruzione mostrati in precedenza, usa la funzione
from_listdi SparsePauliOp per definire gli operatori di Pauli , , . Questo mapperà gli operatori fermionici di creazione e annichilazione sugli operatori di spin dei qubit, codificando il numero di occupazione fermionico nella base computazionale dei qubit. - Genera l'Hamiltoniano desiderato nella base di Pauli applicando gli operatori di cui sopra agli orbitali di interesse. Di solito questo corrisponde alla creazione di una matrice identità che rappresenta gli orbitali del nucleo (non interagenti) e poi all'applicazione degli operatori di creazione e annichilazione allo spazio attivo, con pesi che corrispondono alle specifiche del problema in questione.
Ora che comprendiamo pienamente lo schema di mappatura di Jordan-Wigner, vediamo un esempio più complicato. Potresti ricordare il paper intitolato "Scalable Circuits for Preparing Ground States on Digital Quantum Computers: The Schwinger Model Vacuum on 100 Qubits" della lezione precedente. Non rianalizzeremo il paper in dettaglio — ci concentreremo solo sulla mappatura di Jordan-Wigner, che viene usata per esprimere l'Hamiltoniano di siti di reticolo con siti per il modello di Schwinger in assenza di un campo elettrico.
Qui è molto più difficile identificare precisamente cosa rappresenta un singolo qubit in questo modello, perché è solo l'insieme dei qubit insieme che forma qualcosa di fisico, in questo caso un pacchetto d'onda. Puoi invece pensare approssimativamente ai qubit come a pezzi discretizzati di spazio. Qui è il volume del reticolo in cui ogni elemento (cella unitaria) comprende due qubit. Gli operatori fermionici che abbiamo visto nella slide precedente descrivono l'ampiezza della funzione d'onda su un sito particolare. Quindi il nostro Hamiltoniano contiene questi operatori fermionici di creazione e annichilazione. Usiamo pertanto la trasformazione di Jordan-Wigner per mappare questi operatori negli operatori di Pauli.
dove è l'operatore di Pauli e è Una volta che abbiamo un Hamiltoniano scritto in questo formato, la parte difficile della fase di mapping è terminata, e ora può essere facilmente scritto in un circuito con operatori di Pauli.
Conclusione
Abbiamo discusso quattro esempi di come specifiche tecniche di mapping siano state usate di recente nel campo del quantum computing, partendo dal più semplice e arrivando all'applicazione della trasformazione di Jordan-Wigner alla dinamica adronica. Gran parte di questo materiale era molto tecnica e, se non l'hai mai incontrata prima, può sembrare molto intimidante. Ma diventa più facile con il tempo che si dedica alla pratica — ed è per questo che questo corso si chiama Quantum Computing in Practice. Non è qualcosa che chiunque può semplicemente raccogliere e portare avanti dall'inizio — richiede studio, qualche grattata di testa, e probabilmente momenti di frustrazione. Ma ti incoraggio a restare con quell'disagio e a esplorare davvero le domande che sorgono mentre vai avanti. È l'unico modo per imparare.