Variational Quantum Eigensolver (VQE)
Per questo modulo, gli studenti devono disporre di un ambiente Python funzionante e delle versioni più recenti dei seguenti pacchetti installati:
qiskitqiskit_ibm_runtimeqiskit-aerqiskit.visualizationnumpypylatexenc
Per configurare e installare questi pacchetti, consulta la guida Installa Qiskit. Per eseguire job su computer quantistici reali, gli studenti dovranno configurare un account IBM Cloud, seguendo i passaggi nella guida Configura il tuo account IBM Cloud.
Questo modulo è stato testato e ha utilizzato circa 8 minuti di tempo QPU. Si tratta di una stima e l'utilizzo effettivo può variare.
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-aer qiskit-ibm-runtime scipy
# Uncomment and modify this line as needed to install dependencies
#!pip install 'qiskit>=2.1.0' 'qiskit-ibm-runtime>=0.40.1' 'qiskit-aer>=0.17.0' 'numpy' 'pylatexenc'
Introduzione
Fin dallo sviluppo del modello quantomeccanico all'inizio del XX secolo, gli scienziati hanno capito che gli elettroni non seguono percorsi fissi attorno al nucleo di un atomo, ma esistono piuttosto in regioni di probabilità chiamate orbitali. Questi orbitali corrispondono a livelli energetici specifici e discreti che gli elettroni possono occupare. Gli elettroni risiedono naturalmente nei livelli energetici più bassi disponibili, noti come stato fondamentale. Tuttavia, se un elettrone assorbe energia sufficiente, può saltare a un livello energetico più alto, entrando in uno stato eccitato. Questo stato eccitato è temporaneo e l'elettrone alla fine ritornerà a un livello energetico inferiore, rilasciando l'energia assorbita, spesso sotto forma di luce. Questo processo fondamentale di assorbimento ed emissione di energia è importante per capire come gli atomi interagiscono e formano legami.
Quando gli atomi si uniscono per formare molecole, i loro orbitali atomici si combinano per formare orbitali molecolari. La disposizione e i livelli energetici degli elettroni all'interno di questi orbitali molecolari determinano le proprietà della molecola risultante e la resistenza dei legami chimici. Ad esempio, nella formazione di una molecola di idrogeno () da due atomi di idrogeno individuali, l'elettrone di ciascun atomo occupa orbitali atomici. Quando gli atomi si avvicinano, questi orbitali atomici si sovrappongono e si combinano per formare nuovi orbitali molecolari — uno con energia inferiore (un orbitale di legame) e uno con energia superiore (un orbitale antilegame). I due elettroni, uno da ciascun atomo di idrogeno, occuperanno preferenzialmente l'orbitale di legame a energia inferiore, portando alla formazione di un legame covalente stabile che tiene unita la molecola . La differenza di energia tra gli atomi separati e la molecola formata, in particolare l'energia degli elettroni negli orbitali molecolari, determina la stabilità e le proprietà del legame.
Nelle sezioni seguenti, esploreremo questo processo di formazione molecolare, concentrandoci sulla molecola . Utilizzeremo un vero computer quantistico, combinato con tecniche di ottimizzazione classiche, per trovare l'energia di questo processo semplice ma fondamentale. Questo esperimento fornirà una dimostrazione pratica di come il calcolo quantistico possa essere applicato per risolvere problemi di chimica computazionale, offrendo approfondimenti sul ruolo dell'energia degli elettroni.
VQE - Un algoritmo quantistico variazionale per problemi agli autovalori
Tecniche di approssimazione per la chimica - principio variazionale e set di basi
I contributi di Erwin Schrödinger alla meccanica quantistica non si limitano all'introduzione di un nuovo modello elettronico; fondamentalmente, ha stabilito la meccanica ondulatoria sviluppando la famosa equazione di Schrödinger dipendente dal tempo:
Qui, è l'operatore hamiltoniano, che rappresenta l'energia totale del sistema, e è la funzione d'onda che contiene tutte le informazioni sullo stato quantistico del sistema. (Nota: è la derivata totale rispetto al tempo e non includiamo esplicitamente l'autovalore energetico qui.)
Tuttavia, in molte applicazioni pratiche — come determinare i livelli energetici consentiti di atomi e molecole — utilizziamo invece l'equazione di Schrödinger indipendente dal tempo (equazione agli autovalori dell'energia), che si deriva dalla forma dipendente dal tempo assumendo uno stato stazionario. Uno stato stazionario è uno stato quantistico in cui la densità di probabilità di trovare una particella in un dato punto dello spazio non cambia nel tempo.
In questa forma, rappresenta l'autovalore dell'energia corrispondente allo stato quantistico . L'hamiltoniano include vari contributi energetici, come l'energia cinetica di elettroni e nuclei, le forze attrattive tra elettroni e nuclei e le forze repulsive tra gli elettroni.
La risoluzione dell'equazione agli autovalori dell'energia ci consente di calcolare i livelli energetici quantizzati dei sistemi atomici e molecolari. Tuttavia, per le molecole, risolverla esattamente è difficile perché la funzione d'onda , che descrive la distribuzione spaziale degli elettroni, è complessa e ad alta dimensionalità.
Di conseguenza, gli scienziati utilizzano tecniche di approssimazione per ottenere soluzioni pratiche e accurate. In questo lavoro, ci concentreremo su due metodi chiave:
-
Principio variazionale
Questo metodo approssima la funzione d'onda e la regola per avvicinarsi il più possibile all'energia target, di solito l'energia dello stato fondamentale del sistema. L'idea chiave alla base del principio variazionale è semplice:
- Se indoviniamo una funzione d'onda (una "funzione di prova"), l'energia calcolata da essa sarà sempre uguale o superiore all'energia dello stato fondamentale () del sistema.
- Regolando i parametri nella funzione di prova, , possiamo ottenere un'approssimazione sempre migliore dell'energia dello stato fondamentale.
- La sua accuratezza dipende fortemente dalla scelta della funzione d'onda di prova . Una funzione di prova scelta male può portare a una stima dell'energia lontana dall'accuratezza.
-
Approssimazione del set di basi
Il secondo metodo di approssimazione riguarda la fase di costruzione della funzione d'onda — l'approccio del set di basi. In chimica quantistica, risolvere esattamente l'equazione di Schrödinger per le molecole è quasi impossibile. Invece, approssimiamo la funzione d'onda complessa multi-elettronica costruendola a partire da funzioni matematiche più semplici e predefinite. Un set di basi è essenzialmente una raccolta di queste funzioni matematiche note, tipicamente centrate sugli atomi della molecola, che vengono utilizzate come elementi costitutivi per rappresentare la forma e il comportamento degli elettroni nel sistema. Immagina di cercare di ricreare una scultura dettagliata usando solo una raccolta di mattoncini LEGO standard — più tipi e dimensioni di mattoncini hai (più grande è il set di basi), più accuratamente puoi approssimare la forma originale.
Queste funzioni di base sono spesso ispirate alle soluzioni analitiche per sistemi semplici come l'atomo di idrogeno, assumendo forme come funzioni di tipo gaussiano o di Slater, sebbene siano ancora approssimazioni. Invece di lavorare con gli orbitali molecolari completi teoricamente "esatti" ma intrattabili, li esprimiamo come combinazione lineare (una somma con coefficienti) di queste funzioni di base. Questo metodo è noto come approccio della Combinazione Lineare di Orbitali Atomici (LCAO) quando le funzioni di base assomigliano agli orbitali atomici. Ottimizzando i coefficienti in questa combinazione lineare, possiamo trovare la migliore funzione d'onda approssimata e l'energia possibile entro i limiti del set di basi scelto.
- Più funzioni sono incluse nel set di basi, migliore è l'approssimazione, ma questo ha il costo di un maggiore sforzo computazionale.
- Un set di basi piccolo fornisce una stima approssimativa, mentre un set di basi grande fornisce risultati più precisi a scapito di un maggiore dispendio di risorse computazionali.
In sintesi, per rendere fattibili i calcoli e ridurre il costo computazionale, utilizziamo il principio variazionale approssimando la funzione d'onda, il che riduce la complessità computazionale e consente un'ottimizzazione iterativa per minimizzare l'energia. Nel frattempo, l'approccio del set di basi semplifica i calcoli rappresentando gli orbitali atomici come combinazione di funzioni predefinite, piuttosto che risolvere direttamente una funzione d'onda continua.
Verifica la tua comprensione
Considera la funzione d'onda di prova dove è una costante di normalizzazione e è un parametro regolabile.
(a) Normalizza la funzione d'onda di prova determinando tale che .
Answer
Per normalizzare la funzione d'onda di prova data:
Usa l'integrale gaussiano:
imposta e ottieni:
(b) Calcola il valore di aspettazione dell'hamiltoniano dato da dove , che corrisponde a un potenziale dell'oscillatore armonico semplice.
Answer
L'hamiltoniano per un oscillatore armonico è:
Valore di aspettazione dell'energia cinetica
Calcolando la derivata seconda:
Quindi:
Usando i risultati standard degli integrali gaussiani:
Valore di aspettazione dell'energia potenziale
Usando:
otteniamo:
Valore di aspettazione dell'energia totale
(c) Usa il principio variazionale per trovare il ottimale minimizzando .
Answer
Ottimizza per l'energia minima
Differenzia
Risolvendo:
Sostituendo in :
che corrisponde all'esatta energia dello stato fondamentale dell'oscillatore armonico quantistico.
VQE (Variational Quantum Eigensolver)
Il variational quantum eigensolver (VQE) è il metodo principale che utilizzeremo per esplorare il processo e qui daremo un'occhiata a cos'è il VQE e come funziona. Ma prima facciamo una pausa e osserviamo una cosa molto importante attraverso la domanda di verifica.
Verifica la tua comprensione
Se abbiamo già così tante strategie per i problemi di chimica, perché abbiamo bisogno di un computer quantistico? E qual è lo scopo di utilizzare sia computer quantistici che classici insieme?
Answer
Il calcolo quantistico ha la possibilità di rivoluzionare la chimica affrontando problemi con cui i computer classici faticano a causa della scalabilità esponenziale degli stati quantistici. Richard Feynman ha notoriamente osservato che per simulare la natura, i calcoli devono essere anch'essi quantistici [ref 1].
Ad esempio, simulare la caffeina con il set di basi più semplice (STO-3G) richiederebbe bit, molto più grande del numero totale di stelle nell'universo osservabile () [ref 2]. Un computer quantistico può descrivere gli orbitali elettronici della caffeina con 160 qubit.
I computer quantistici elaborano naturalmente le interazioni quantistiche usando la sovrapposizione e l'entanglement, che forniscono un modo promettente per consentire simulazioni molecolari accurate. Inoltre, possiamo combinare i vantaggi sia dei computer quantistici (simulazione degli elettroni) che dei computer classici (pre/post-elaborazione dei dati, gestione del processo algoritmico, ottimizzazione e così via). Questi sono attesi per migliorare la scoperta di materiali, la progettazione di farmaci e le previsioni di reazione, riducendo costosi esperimenti per tentativi ed errori. [ref 3][ref 4]
Se vuoi sapere perché i computer quantistici sono necessari per i problemi di chimica e perché usare sia risorse di calcolo quantistico che classico, consulta i seguenti articoli:
Ora torniamo al VQE.
Il VQE combina la potenza dei computer quantistici con i computer classici, utilizzando fondamentalmente principi variazionali per ottenere l'energia dello stato fondamentale del sistema. Per capire il VQE, scomponilo prima in tre parti:
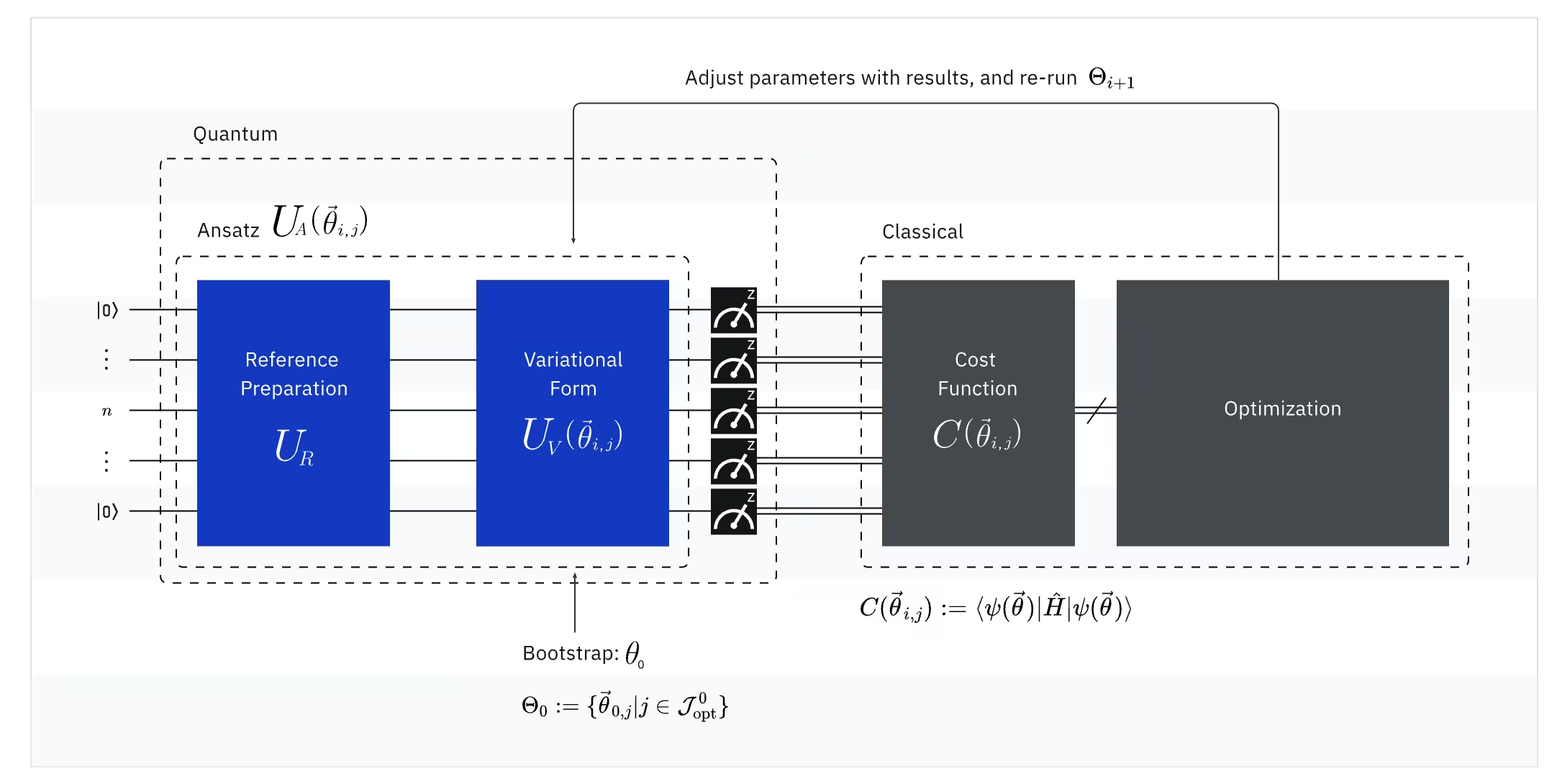
(Quantistico) Observable: l'hamiltoniano molecolare (energia di una molecola)
Nel VQE, l'hamiltoniano molecolare/atomico è un observable, il che significa che possiamo misurare il suo valore attraverso un esperimento. Il nostro obiettivo è trovare l'energia più bassa possibile (l'energia dello stato fondamentale) della molecola. Per fare questo, utilizziamo uno stato quantistico di prova, generato da un circuito quantistico parametrizzato (ansatz). Misuriamo l'observable e ottimizziamo lo stato quantistico finché non raggiungiamo l'energia più bassa possibile.
Il set di basi utilizzato per l'hamiltoniano molecolare determina il numero di qubit necessari e influisce direttamente sull'accuratezza del VQE. Scegliere il set di basi giusto è fondamentale per bilanciare efficienza e precisione. Per semplificare i calcoli senza cambiare il set di basi, possiamo usare strategie come l'imposizione della simmetria e la riduzione dello spazio attivo. Molte molecole hanno forme simmetriche (come una farfalla o un fiocco di neve), il che significa che alcune parti si comportano allo stesso modo. Invece di calcolare tutto separatamente, possiamo concentrarci solo sulle parti uniche, risparmiando risorse quantistiche, sfruttando così la simmetria. Nella riduzione dello spazio attivo, consideriamo solo gli orbitali importanti, poiché non tutti gli elettroni influenzano significativamente l'energia molecolare. Gli elettroni vicini al nucleo rimangono per lo più invariati, mentre altri influenzano il legame. Applicando questi metodi, possiamo rendere il VQE più efficiente mantenendo l'accuratezza.
Una volta ottenuto un hamiltoniano molecolare usando il set di basi e le strategie appropriate di cui sopra, dobbiamo trasformare questo hamiltoniano in uno adatto ai computer quantistici. La mappatura dei problemi sugli operatori di Pauli può essere piuttosto complicata. Questo è particolarmente vero in chimica quantistica, che lavora con particelle indistinguibili (elettroni), poiché i qubit sono distinguibili. Non entreremo nei dettagli delle mappature qui, ma ti rimandiamo alle seguenti risorse. Una discussione generale sulla mappatura di un problema su operatori quantistici si trova in Quantum computing in practice. Una discussione più dettagliata sulla mappatura di problemi di chimica in operatori quantistici si trova in Quantum chemistry with VQE.
Per questo modulo, ti forniremo gli hamiltoniani (a un qubit) appropriati per e in modo da poterci concentrare sull'utilizzo del computer quantistico. Questi hamiltoniani a un qubit sono preparati usando il set di basi STO-6G e la mappatura di Jordan-Wigner, che è la mappatura più diretta con la più semplice interpretazione fisica, perché mappa l'occupazione di un spin-orbitale all'occupazione di un qubit. Abbiamo anche usato una tecnica di riduzione dei qubit usando una simmetria dell'hamiltoniano, che usa i pattern nel comportamento delle occupazioni di spin per ridurre il numero di qubit. Per la molecola , assumiamo che la distanza tra i due atomi di idrogeno sia 0.735 .
(Quantistico) Ansatz: la funzione d'onda di prova (Come costruire uno stato quantistico banale con un circuito quantistico)
Per il VQE, l'ansatz (plurale: ansätze) è composto da due componenti chiave. Il primo è la preparazione dello stato iniziale, che imposta lo stato del qubit applicando gate quantistici senza parametro variazionale. Il secondo componente è il circuito quantistico parametrizzato, un circuito quantistico speciale con parametri regolabili, simile alle manopole di una radio. Questi parametri verranno utilizzati per l'ultima parte — l'ottimizzatore classico — per aiutarci a raggiungere il miglior stato fondamentale possibile.
Nella sezione sul principio variazionale, abbiamo imparato che la qualità dello stato di prova influisce sulla qualità dei risultati dell'algoritmo variazionale. Ciò significa che scegliere un buon ansatz è importante nel VQE. Ancora una volta, questo è un argomento ricco e complesso. Non tratteremo qui i diversi tipi di ansatz o le loro origini. Se sei interessato a saperne di più sui circuiti quantistici parametrizzati e l'ansatz, puoi esplorare la lezione Ansatz and variational form del corso Variational algorithm design, che fornisce spiegazioni dettagliate ed esempi di ansätze.
Poiché utilizzeremo un hamiltoniano a un qubit in questo modulo, abbiamo bisogno di un circuito quantistico parametrizzato a un qubit come ansatz. Vedremo tre tipi di ansätze a un qubit nella sezione seguente. Li confronteremo e discuteremo le considerazioni chiave nella scelta di un ansatz.
(Classico) Ottimizzatore: ottimizzazione del circuito quantistico
Una volta che il computer quantistico misura l'energia dell'observable dall'ansatz, i parametri dell'ansatz e il valore dell'energia vengono inviati all'ottimizzatore classico per la regolazione. Questo processo di ottimizzazione viene eseguito su un computer classico, tipicamente usando pacchetti scientifici di uso generale come SciPy.
L'ottimizzatore classico tratta l'energia misurata come una funzione di costo. Nei problemi di ottimizzazione, una funzione di costo (a volte chiamata anche funzione obiettivo) è una funzione matematica che misura quanto "buona" è una particolare soluzione. L'obiettivo dell'ottimizzatore è trovare l'insieme di parametri che minimizza questa funzione di costo. Nel contesto della ricerca dell'energia dello stato fondamentale di una molecola, l'energia stessa funge da funzione di costo — vogliamo trovare i parametri per il nostro circuito quantistico (la nostra "soluzione") che producono l'energia più bassa possibile. L'ottimizzatore classico usa questo valore di energia misurato (il costo) e determina il prossimo insieme di parametri ottimizzati per l'ansatz quantistico. Questi parametri aggiornati vengono poi inviati nuovamente al circuito quantistico e il processo si ripete. Ad ogni iterazione, l'ottimizzatore classico regola i parametri cercando di ridurre l'energia (minimizzare la funzione di costo) finché non viene soddisfatto un criterio di convergenza predefinito, assicurando idealmente che venga trovata l'energia più bassa possibile (corrispondente allo stato fondamentale della molecola per quella distanza di legame e quel set di basi).
Ci sono molte strategie di ottimizzazione fornite da pacchetti scientifici come SciPy. Puoi trovare ulteriori informazioni nella lezione Optimization loops del corso Variational algorithm design. Qui utilizzeremo COBYLA (Constrained Optimization BY Linear Approximations), un algoritmo di ottimizzazione adatto a paesaggi energetici complicati. In particolare, COBYLA non tenta di calcolare un gradiente della funzione studiata; questo viene chiamato ottimizzatore privo di gradiente. Immagina di cercare di trovare il picco più alto in una catena montuosa con gli occhi chiusi. Poiché non puoi vedere l'intero paesaggio, fai piccoli passi in diverse direzioni, controllando se stai salendo o scendendo. COBYLA funziona in modo simile — si muove attraverso lo spazio dei parametri, testando valori diversi, migliorando gradualmente il risultato finché non trova il migliore.
Ora sei pronto per effettuare un calcolo VQE. A tal fine, prova la domanda di verifica di seguito, che riepiloga il processo complessivo.
Verifica la tua comprensione
Riempi i vuoti con i termini corretti per completare il riepilogo del processo VQE, poi clicca per verificare le tue risposte.
Il VQE è un algoritmo quantistico variazionale, che combina la potenza del (1) ________ e del calcolo classico, utilizzato per trovare il (2) __________ di una molecola. Il processo inizia definendo il (3) __________, che rappresenta l'energia totale del sistema e agisce come observable nelle misurazioni quantistiche. Successivamente, prepariamo un (4) __________, un circuito quantistico con parametri regolabili che rappresenta la funzione d'onda di prova della molecola. Questi parametri vengono ottimizzati usando un (5) __________, un algoritmo classico che regola i parametri iterativamente per minimizzare l'energia misurata. Nella discussione sopra abbiamo usato l'ottimizzatore (6) __________, che raffina i parametri dell'ansatz senza necessità di calcoli di derivate. Il processo continua finché non raggiungiamo la (7) __________, il che significa che abbiamo trovato l'energia più bassa possibile della molecola.
Banca delle parole:
- classical optimizer
- ground state energy
- hardware-efficient
- ansatz
- molecular Hamiltonian
- COBYLA
- quantum computing
- convergence
Answer
1 → quantum computing
2 → ground state energy
3 → molecular Hamiltonian
4 → ansatz
5 → classical optimizer
6 → COBYLA
7 → convergence
Calcola l'energia dello stato fondamentale di un atomo di idrogeno con VQE
Ora usiamo quello che abbiamo imparato per calcolare l'energia dello stato fondamentale di un atomo di idrogeno. Nel corso del modulo, utilizzeremo un framework per il calcolo quantistico noto come "Qiskit patterns", che suddivide i workflow nei seguenti passi:
- Passo 1: Mappare gli input classici in un problema quantistico
- Passo 2: Ottimizzare il problema per l'esecuzione quantistica
- Passo 3: Eseguire usando le primitive Qiskit Runtime
- Passo 4: Post-elaborazione e analisi classica

In generale seguiremo questi passi.
Iniziamo caricando alcuni pacchetti necessari, incluse le primitive Qiskit Runtime. Selezioneremo anche il computer quantistico meno occupato disponibile.
Il codice qui sotto consente di salvare le credenziali al primo utilizzo. Assicurati di eliminare queste informazioni dal notebook dopo averle salvate nel tuo ambiente, in modo che le tue credenziali non vengano condivise accidentalmente quando condividi il notebook. Consulta Configura il tuo account IBM Cloud e Inizializza il servizio in un ambiente non attendibile per ulteriori indicazioni.
# Load the Qiskit Runtime service
from qiskit_ibm_runtime import QiskitRuntimeService
# Load the Runtime primitive and session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# Syntax for first saving your token. Delete these lines after saving your credentials.
# QiskitRuntimeService.save_account(channel='ibm_quantum_platform',
# instance = '<YOUR_IBM_INSTANCE_CRN>', token='<YOUR-API_KEY>', overwrite=True, set_as_default=True)
# service = QiskitRuntimeService(channel='ibm_quantum_platform')
# Load saved credentials
service = QiskitRuntimeService()
# Use the least busy backend, or uncomment the loading of a specific backend like "ibm_brisbane".
backend = service.least_busy(operational=True, simulator=False, min_num_qubits=127)
# backend = service.backend("ibm_brisbane")
print(backend.name)
ibm_brisbane
La cella qui sotto ti consentirà di passare dall'uso del simulatore all'hardware reale per tutto il notebook. Ti consigliamo di eseguirla ora:
# Load the Aer simulator and generate a noise model based on the currently-selected backend.
from qiskit_aer import AerSimulator
from qiskit_aer.noise import NoiseModel
# Alternatively, load a fake backend with generic properties and define a simulator.
noise_model = NoiseModel.from_backend(backend)
# Define a simulator using Aer, and use it in Sampler.
backend_sim = AerSimulator(noise_model=noise_model)
Passo 1: Mappare il problema in circuiti e operatori quantistici
Avviamo il calcolo VQE definendo l'Hamiltoniano per la molecola di idrogeno () a una specifica distanza di legame. Questo Hamiltoniano rappresenta l'energia totale del sistema in termini di operatori su qubit, prodotto e mappato dal sistema molecolare tramite una procedura standard: 1) impiegando il set di basi STO-6G (una specifica raccolta di funzioni matematiche usate per approssimare gli orbitali elettronici), 2) applicando la mappatura Jordan-Wigner (una tecnica per tradurre gli operatori fermionici che descrivono gli elettroni in operatori su qubit), e 3) eseguendo la riduzione di qubit usando la parità dell'Hamiltoniano per semplificare il problema.
Come spiegato in precedenza, le energie dello stato fondamentale calcolate dipendono fortemente dalla scelta del set di basi e dalla geometria molecolare (come la distanza di legame). Per questa configurazione specifica e dopo queste trasformazioni, l'Hamiltoniano su qubit risultante è semplice:
Qui, rappresenta l'operatore identità e rappresenta l'operatore di Pauli-Z, che agisce su un singolo qubit. I coefficienti derivano dagli integrali calcolati usando il set di basi STO-6G a questa particolare distanza di legame con la trasformazione appropriata.
Con questo Hamiltoniano definito, possiamo ora usare VQE per calcolare la sua energia dello stato fondamentale. È utile confrontare l'energia dello stato fondamentale calcolata con i valori attesi. Per un singolo atomo di idrogeno (H) isolato, l'energia dello stato fondamentale è esattamente -0,5 Hartree (in assenza di effetti relativistici). Calcoliamo l'energia esatta dello stato fondamentale del nostro specifico Hamiltoniano su qubit come definito sopra e confrontiamola con i valori noti pertinenti.
from qiskit.quantum_info import SparsePauliOp
import numpy as np
# Qubit Hamiltonian of the hydrogen atom generated by using STO-3G basis set and parity mapping
Hamiltonian = SparsePauliOp.from_list([("I", -0.2355), ("Z", 0.2355)])
# exact ground state energy of Hamiltonian
A = np.array(Hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print(
"The exact ground state energy of the Hamiltonian is ",
min(eigenvalues).real,
"hartree",
)
h = min(eigenvalues.real)
The exact ground state energy of the Hamiltonian is -0.471 hartree
Successivamente, abbiamo bisogno di un circuito quantistico parametrizzato, un ansatz, per preparare una funzione d'onda di prova per lo stato fondamentale. L'obiettivo è trovare i parametri che minimizzano il valore di aspettazione dell'energia . La scelta dell'ansatz è cruciale perché determina l'insieme degli stati quantistici possibili che il nostro circuito può preparare. Un "buon" ansatz è uno sufficientemente flessibile da rappresentare uno stato molto vicino al vero stato fondamentale dell'Hamiltoniano che stiamo studiando, ma non così complesso da richiedere troppi parametri o un circuito troppo profondo per i computer quantistici attuali.
Qui proveremo tre diversi ansätze a singolo qubit per vedere quale fornisce una migliore "copertura" dei possibili stati quantistici in cui un singolo qubit può trovarsi. La "copertura" si riferisce all'insieme degli stati quantistici che il circuito ansatz può produrre variando i suoi parametri.
Useremo tre ansätze basati su diverse combinazioni di gate di rotazione a singolo qubit:
- Un ansatz con gate di rotazione su 1 asse: questo ansatz usa rotazioni attorno a un singolo asse (). Sulla sfera di Bloch, ciò corrisponde a muoversi solo lungo un cerchio specifico. Questo è il meno flessibile e copre un insieme limitato di stati.
- Due ansätze con gate di rotazione su 2 assi: questi ansätze combinano rotazioni attorno a due assi diversi ( e ). Ciò consente di raggiungere una porzione più ampia della sfera di Bloch rispetto a una rotazione su asse singolo.
Confrontando i risultati VQE ottenuti con questi tre ansätze, possiamo vedere come la flessibilità e la copertura dello spazio degli stati dell'ansatz influenzino la nostra capacità di trovare la vera energia dello stato fondamentale del nostro Hamiltoniano semplificato. Un ansatz più flessibile ha il potenziale di trovare un'approssimazione migliore, ma potrebbe anche essere più difficile da gestire per l'ottimizzatore classico.
from qiskit import QuantumCircuit
from qiskit.circuit import Parameter
from qiskit.quantum_info import Statevector, DensityMatrix, Pauli
theta = Parameter("θ")
phi = Parameter("φ")
lam = Parameter("λ")
ansatz1 = QuantumCircuit(1)
ansatz1.rx(theta, 0)
ansatz2 = QuantumCircuit(1)
ansatz2.rx(theta, 0)
ansatz2.rz(phi, 0)
ansatz3 = QuantumCircuit(1)
ansatz3.rx(theta, 0)
ansatz3.rz(phi, 0)
ansatz3.rx(lam, 0)
<qiskit.circuit.instructionset.InstructionSet at 0x1059def80>
Ora generiamo 5000 numeri casuali per ogni parametro e tracciamo la distribuzione degli stati quantistici casuali, generati dai tre ansätze con questi parametri casuali. Puoi pensare a questi parametri come rotazioni attorno a diversi assi su una superficie sferica. Per visualizzare la distribuzione degli stati quantistici, useremo la sfera di Bloch, una sfera tridimensionale che mostra lo stato di un singolo qubit. Qualsiasi punto sulla sfera rappresenta un possibile stato del qubit, dove i poli nord e sud sono come il classico "0" e "1", ma il qubit può trovarsi anche in qualsiasi punto intermedio, mostrando proprietà quantistiche speciali come la sovrapposizione. Per prima cosa, prepara le funzioni necessarie per tracciare la sfera di Bloch 3D e prepara 5000 parametri casuali.
import matplotlib.pyplot as plt
def plot_bloch(bloch_vectors):
# Extract X, Y, Z coordinates for 3D projection
X_coords = bloch_vectors[:, 0]
Z_coords = bloch_vectors[:, 2]
# Compute Y coordinates from X and Z to approximate the full Bloch sphere projection
Y_coords = bloch_vectors[:, 1]
# Create 3D plot
fig = plt.figure(figsize=(8, 8))
ax = fig.add_subplot(111, projection="3d")
ax.scatter(X_coords, Y_coords, Z_coords, color="blue", alpha=0.6)
# Labels and title
ax.set_xlabel("X")
ax.set_ylabel("Y")
ax.set_zlabel("Z")
ax.set_title("Parameterized 1-Qubit Circuit on 3D Bloch Sphere")
# Set axis limits and make them equal
ax.set_xlim([-1, 1])
ax.set_ylim([-1, 1])
ax.set_zlim([-1, 1])
# Ensure equal aspect ratio for all axes
ax.set_box_aspect([1, 1, 1]) # Equal scaling for x, y, z axes
# Show grid
ax.grid(True)
plt.show()
num_samples = 5000 # Number of random states
theta_vals = np.random.uniform(0, 2 * np.pi, num_samples)
phi_vals = np.random.uniform(0, 2 * np.pi, num_samples)
lam_vals = np.random.uniform(0, 2 * np.pi, num_samples)
Vediamo come funziona il nostro primo ansatz.
# List to store Bloch Sphere XZ coordinates
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create a circuit and bind parameters
qc = ansatz1
bound_qc = qc.assign_parameters({theta: theta_vals[i]}) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to a numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
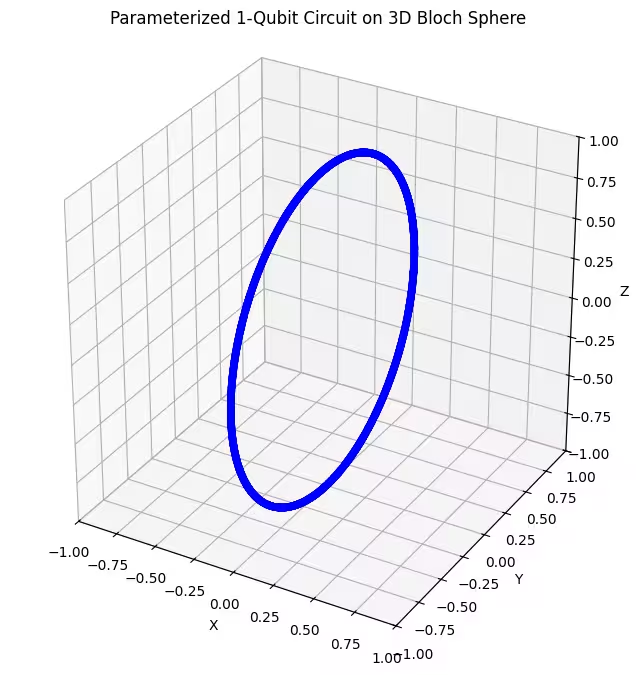
Possiamo vedere che il nostro primo ansatz restituisce stati quantistici distribuiti a forma di anello sulla sfera di Bloch. Questo ha senso, perché abbiamo fornito all'ansatz un solo parametro di rotazione. Può quindi produrre solo stati ruotati attorno a un asse. Partendo dal punto e ruotando attorno a un asse si ottiene sempre un anello. Controlliamo ora il nostro secondo ansatz, che ha due gate di rotazione ortogonali - Rx e Rz.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz2
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i]}
) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
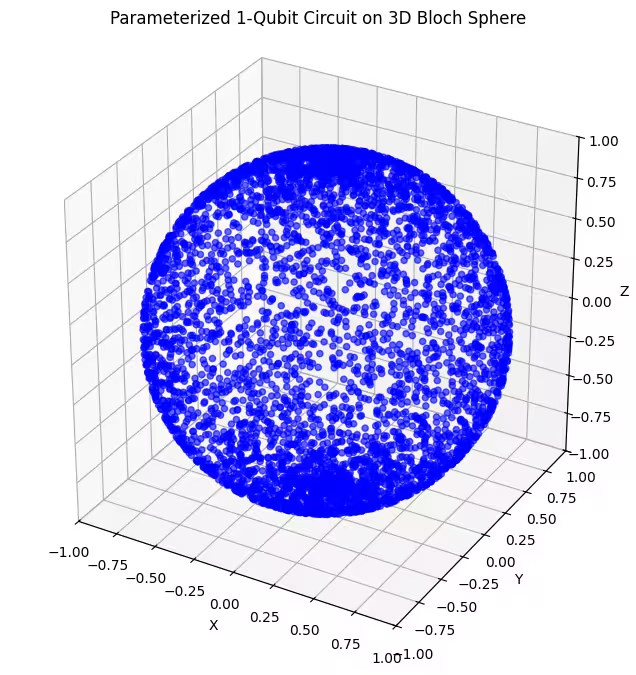
Qui possiamo vedere che il nostro secondo ansatz copre una porzione più ampia della sfera di Bloch — nota però che i punti sono più concentrati attorno ai poli e più dispersi attorno all'equatore. Ora è il momento di controllare il nostro ultimo ansatz.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz3
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i], lam: lam_vals[i]}
)
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
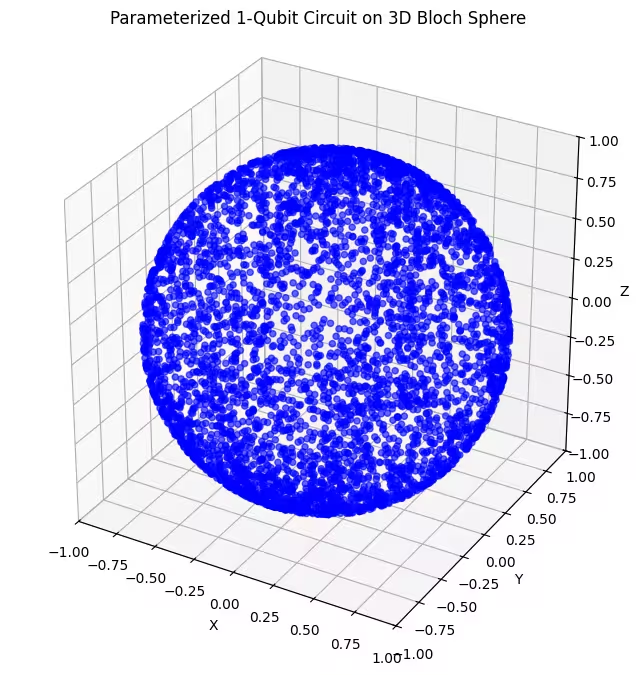
Qui puoi vedere gli stati quantistici distribuiti in modo più uniforme generati dal nostro ultimo ansatz.
Come accennato, la cosa migliore da fare è acquisire conoscenza dello stato fondamentale che si cerca e usare un ansatz adatto a sondare gli stati vicini a quello stato fondamentale. Per esempio, se sapessimo che il nostro stato fondamentale si trova vicino a un polo, potremmo selezionare l'ansatz 2. Per semplicità, continueremo con l'ansatz 3, che sonda uniformemente l'intera sfera di Bloch.
Ora che abbiamo selezionato il nostro ansatz, tracciamo il circuito.
# Pre-defined ansatz circuit and operator class for Hamiltonian
ansatz = ansatz3
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.draw("mpl", style="iqp")
This circuit has 3 parameters

Passo 2: Ottimizzare per l'hardware di destinazione
Quando si esegue un calcolo su un vero computer quantistico, non ci interessa solo la logica del circuito quantistico. Ci interessano anche cose come quali operazioni possono essere eseguite da quel particolare computer quantistico, e dove su di esso si trovano i qubit che stiamo usando. Sono vicini tra loro? Sono lontani? Pertanto, il passo successivo consiste nel riscrivere il nostro circuito usando gate che siano naturali per il computer quantistico che utilizzeremo, tenendo conto del layout dei qubit. Questo può essere fatto tramite la transpilazione — dopo questo processo, puoi vedere il nostro semplice ansatz convertito in un diverso insieme di gate, e i nostri qubit astratti saranno mappati in qubit fisici su un vero computer quantistico.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
config = backend.configuration()
print("Backend: {config.backend_name}")
print("Native gates: ", config.supported_instructions, ",")
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")
Backend: {config.backend_name}
Native gates: ['ecr', 'id', 'delay', 'measure', 'reset', 'rz', 'sx', 'x'] ,

Puoi vedere che i gate rx, rz del nostro ansatz sono stati convertiti in una serie di gate rz, sx, che sono i gate nativi del nostro backend. Inoltre, puoi vedere che il nostro q0 è ora mappato nel quinto qubit fisico. Dobbiamo anche mappare il nostro Hamiltoniano secondo queste modifiche, come nel codice seguente:
Hamiltonian_isa = Hamiltonian.apply_layout(layout=ansatz_isa.layout)
Passo 3: Eseguire sull'hardware di destinazione
Ora è il momento di eseguire il nostro VQE su un vero QPU. Per questo, abbiamo prima bisogno di una funzione di costo per il processo di ottimizzazione, che valuta il valore di aspettazione dell'Hamiltoniano con uno stato quantistico generato dall'ansatz. Non preoccuparti! Non devi scrivere tutto il codice da solo. Abbiamo preparato una funzione per questo, e tutto quello che devi fare è eseguire la cella qui sotto.
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
Infine, prepariamo i parametri iniziali per il nostro ansatz e il suo processo di ottimizzazione. Puoi semplicemente usare tutti zeri o valori casuali. Abbiamo selezionato i parametri iniziali qui sotto, ma sentiti libero di commentare o decommentare le righe nella cella per campionare i parametri in modo casuale, uniformemente da 0 a .
# x0 = np.random.uniform(0, 2*pi, 3)
x0 = [1, 1, 0]
# QPU Est. 2min for ibm_brisbane
from scipy.optimize import minimize
from qiskit_ibm_runtime import Batch
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, Hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 10, "tol": 0.01},
)
batch.close()
Iters. done: 1 [Current cost: -0.3361517318448143]
Iters. done: 2 [Current cost: -0.4682546422099432]
Iters. done: 3 [Current cost: -0.38985802144149584]
Iters. done: 4 [Current cost: -0.38319217316749354]
Iters. done: 5 [Current cost: -0.4628720756579032]
Iters. done: 6 [Current cost: -0.4683301936226905]
Iters. done: 7 [Current cost: -0.45480498699294747]
Iters. done: 8 [Current cost: -0.4690533242050814]
Iters. done: 9 [Current cost: -0.465867415110354]
Iters. done: 10 [Current cost: -0.4606882723137227]
h_vqe = res.fun
print("The reference ground state energy is ", min(eigenvalues))
print("The computed ground state energy is ", h_vqe)
The reference ground state energy is (-0.471+0j)
The computed ground state energy is -0.4690533242050814
Congratulazioni! Hai appena completato con successo il tuo primo esperimento di chimica quantistica. Possiamo vedere una differenza tra l'energia esatta dello stato fondamentale dell'Hamiltoniano e la nostra, ma poiché abbiamo usato una tecnica di mitigazione degli errori predefinita (che corregge gli errori di lettura), la differenza è minima. Questo è un ottimo inizio!
Nota: puoi ottenere un risultato migliore impostando un livello di mitigazione degli errori usando resilience_level. Il valore predefinito è 1, e se imposti un valore più alto, verrà utilizzato più tempo QPU ma potrebbe restituire un risultato migliore.
Passo 4: Post-elaborazione
È il momento di osservare come ha lavorato il nostro ottimizzatore classico. Esegui la cella qui sotto e osserva il pattern di convergenza.
fig, ax = plt.subplots()
x = np.linspace(0, 10, 10)
# Define the constant function
y_constant = np.full_like(x, h)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

Abbiamo iniziato con un valore iniziale abbastanza buono, tale da ottenere un buon valore finale in soli 10 passi. Puoi vedere picchi grandi e piccoli, e questa è la caratteristica tipica dell'ottimizzatore COBYLA — esplora lo spazio come se non potesse vedere il paesaggio e regola la dimensione dei passi ad ogni misurazione.
Verifica la tua comprensione
Qual è la tua osservazione? Quale parte del processo descritto sopra è aperta a miglioramenti per ottenere risultati più vicini ai valori teorici, o più vicini all'energia precisa dello stato fondamentale dell'Hamiltoniano? Quali sono alcune cose da considerare?
Answer
La prima cosa da considerare è il cambiamento dell'insieme di basi usato nel calcolo dell'Hamiltoniano delle molecole. Come accennato in precedenza, l'energia dello stato fondamentale dell'atomo H è -0,5 Hartree, come è ben noto, e la base STO-6G che abbiamo scelto non è sufficiente per derivare questo valore con precisione.
La scelta di un tipo di base più complessa aumenta il numero di qubit usati dall'Hamiltoniano; pertanto, dobbiamo selezionare un ansatz più complesso e adatto per i problemi di chimica.
Il prossimo da ottimizzare è la gestione del rumore nel QPU. Tecniche di mitigazione degli errori più avanzate producono risultati migliori ma potrebbero richiedere più tempo. Considera inoltre come il shot_number influisce sui risultati.
Infine, si può ottenere anche una migliore performance di convergenza provando ottimizzatori diversi.
Calcola l'energia dello stato fondamentale della molecola di idrogeno con VQE
Ora che abbiamo esaminato il processo complessivo di VQE usando atomi di , calcoleremo l'energia dello stato fondamentale della molecola in modo più rapido.
Passo 1: Mappa il problema su circuiti quantistici e operatori
Qui ti forniamo anche un Hamiltoniano a un qubit che utilizza la base STO-6G e la trasformazione di Jordan-Wigner, con riduzione dei qubit tramite una simmetria dell'Hamiltoniano. Nota che abbiamo usato una distanza atomica tra i due atomi di idrogeno di 0.735 .
A differenza del calcolo di un singolo atomo di idrogeno (), per calcolare lo stato fondamentale di una molecola di idrogeno (), dobbiamo considerare anche la forza repulsiva tra i nuclei dei due atomi di idrogeno, oltre all'energia associata agli orbitali elettronici. In questo passo forniremo questo valore come costante, e lo calcoleremo effettivamente nel problema di verifica intermedia.
h2_hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
# exact ground state energy of hamiltonian
nuclear_repulsion = 0.71997
A = np.array(h2_hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print("Electronic ground state energy (Hartree): ", min(eigenvalues).real)
print("Nuclear repulsion energy (Hartree): ", nuclear_repulsion)
print(
"Total ground state energy (Hartree): ", min(eigenvalues).real + nuclear_repulsion
)
h2 = min(eigenvalues).real + nuclear_repulsion
Electronic ground state energy (Hartree): -1.8659468547627318
Nuclear repulsion energy (Hartree): 0.71997
Total ground state energy (Hartree): -1.1459768547627318
Passo 2: Ottimizza per l'hardware di destinazione
Poiché il numero di qubit usati dal VQE e dall'Hamiltoniano precedenti è lo stesso del backend da usare per l'esecuzione, utilizzeremo l'ansatz esistente e la sua forma ottimizzata.
h2_hamiltonian_isa = h2_hamiltonian.apply_layout(layout=ansatz_isa.layout)
Passo 3: Esegui sull'hardware di destinazione
Ora è il momento di eseguire i calcoli sul QPU reale. Quasi tutto è identico, ma useremo il punto iniziale appropriato per adattarci all'Hamiltoniano. Inoltre, nella parte iterativa, alcune impostazioni dell'Estimator, usato per calcolare i valori di aspettazione dell'Hamiltoniano per l'ansatz nel QPU, saranno configurate in modo leggermente diverso rispetto ai calcoli precedenti. Discuteremo questa modifica più in dettaglio in una domanda di verifica intermedia.
x0 = [2, 0, 0]
# QPU time 4min for ibm_brisbane
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, h2_hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 15},
)
batch.close()
Iters. done: 1 [Current cost: -0.710621837568328]
Iters. done: 2 [Current cost: -0.2603208441168329]
Iters. done: 3 [Current cost: -0.25548711201326424]
Iters. done: 4 [Current cost: -0.581129450619904]
Iters. done: 5 [Current cost: -1.722920997605439]
Iters. done: 6 [Current cost: -1.6633324849371915]
Iters. done: 7 [Current cost: -1.8066989598929164]
Iters. done: 8 [Current cost: -1.8051093803839542]
Iters. done: 9 [Current cost: -1.802692217571555]
Iters. done: 10 [Current cost: -1.8233585485263144]
Iters. done: 11 [Current cost: -1.6904116652617205]
Iters. done: 12 [Current cost: -1.8245120321245392]
Iters. done: 13 [Current cost: -1.6837021361383608]
Iters. done: 14 [Current cost: -1.8166632606115467]
Iters. done: 15 [Current cost: -1.863446212658907]
h2_vqe = res.fun + nuclear_repulsion
print(
"The reference ground state energy is ", min(eigenvalues).real + nuclear_repulsion
)
print("The computed ground state energy is ", h2_vqe)
The reference ground state energy is -1.1459768547627318
The computed ground state energy is -1.143476212658907
Sebbene il VQE fornisca teoricamente un limite superiore alla vera energia dello stato fondamentale, le implementazioni pratiche su hardware quantistico reale o simulato con rumore, così come le approssimazioni introdotte nella preparazione dell'Hamiltoniano (come i set di base o la riduzione dei qubit), possono introdurre errori che talvolta portano a un'energia misurata leggermente inferiore al valore teorico esatto o a uno specifico valore di riferimento numerico. Sebbene ci siano alcuni errori, i risultati sembrano essere soddisfacenti, specialmente considerando il numero ridotto di passi. Ora, concludiamo questo calcolo VQE esaminando come ha lavorato l'ottimizzatore.
Passo 4: Post-elaborazione
fig, ax = plt.subplots()
x = np.linspace(0, 5, 15)
# Define the constant function
y_constant = np.full_like(x, min(eigenvalues))
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

Verifica la tua comprensione
Calcoliamo l'energia di repulsione nucleare della molecola , che abbiamo incluso come valore costante (0,71997 Hartree).

Usa la legge di Coulomb e le unità atomiche per assicurarti di ottenere il valore in Hartree.
Answer
Poiché entrambi i nuclei di idrogeno sono carichi positivamente, si respingono a vicenda per effetto della forza elettrostatica. Questa repulsione è descritta dalla legge di Coulomb:
dove è la carica del protone, è la permittività del vuoto, e è la distanza tra i due nuclei, misurata in metri o raggi di Bohr in unità di joule (J).
Per calcolare questa energia in Hartree, è necessario convertire l'equazione sopra nel sistema di Unità Atomiche (AU). In AU, , e il raggio di Bohr () è 1, diventando la scala di lunghezza fondamentale in AU. Con queste semplificazioni, la legge di Coulomb si riduce a:
dove deve essere misurato in raggi di Bohr ().
Per convertire la separazione nucleare data in in , è necessaria questa relazione di conversione:
quindi diventa .
Pertanto, l'energia di repulsione nucleare di un dato è
Calcola l'energia di reazione di
Ora utilizziamo ciò che abbiamo ottenuto! Hai usato VQE, un risolutore variazionale quantistico degli autovalori, per calcolare l'energia dello stato fondamentale dell'atomo e della molecola . Ciò che resta è usare i valori calcolati per ottenere l'energia di reazione del processo .
L'energia di reazione è la variazione di energia che avviene quando le sostanze reagiscono per formare nuove sostanze. Immagina di stare costruendo qualcosa: a volte devi fornire energia (come impilare dei blocchi), e a volte l'energia viene rilasciata (come una palla che rotola in discesa). In chimica, le reazioni possono assorbire energia (endotermiche) o rilasciarla (esotermiche).
L'energia di reazione del processo può essere calcolata con la seguente formula:
Eseguendo la cella qui sotto, vediamo questo visivamente. Useremo il valore esatto dello stato fondamentale di ciascun Hamiltoniano e confronteremo l'energia di reazione della soluzione esatta con i risultati VQE.
# Theoretical values
E_H_theo = h.real
E_H2_theo = h2
# Experimental values
E_H_exp = h_vqe
E_H2_exp = h2_vqe
# Calculate reaction energies
E_reaction_theo = E_H2_theo - (2 * E_H_theo)
E_reaction_exp = E_H2_exp - (2 * E_H_exp)
# Set up the plot
fig, ax = plt.subplots(figsize=(8, 6))
ax.set_xlim(0, 3)
ax.set_ylim(-1.16, -0.93) # Adjust y-axis range to highlight differences
ax.set_xticks([])
ax.set_ylabel("Energy (Hartree)")
ax.set_title("H + H → H₂ Reaction Energy Diagram")
# Plot theoretical energy levels
ax.hlines(
y=2 * E_H_theo, xmin=0.5, xmax=1.3, linewidth=2, color="r", label="2H (Exact)"
)
ax.hlines(y=E_H2_theo, xmin=1.3, xmax=2, linewidth=2, color="b", label="H₂ (Exact)")
# Plot experimental energy levels
ax.hlines(
y=2 * E_H_exp,
xmin=0.5,
xmax=1.5,
linewidth=2,
color="r",
linestyle="dashed",
label="2H (VQE)",
)
ax.hlines(
y=E_H2_exp,
xmin=1.5,
xmax=2.5,
linewidth=2,
color="b",
linestyle="dashed",
label="H₂ (VQE)",
)
# Add labels
ax.text(
1,
2 * E_H_theo,
f"2H: {2*E_H_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
2,
E_H2_theo,
f"H₂: {E_H2_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
1,
2 * E_H_exp,
f"2H_VQE: {2*E_H_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
ax.text(
2,
E_H2_exp,
f"H₂_VQE: {E_H2_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
# Add arrows for reaction energy with ΔE label in the middle
mid_y_theo = (2 * E_H_theo + E_H2_theo) / 2
mid_y_exp = (2 * E_H_exp + E_H2_exp) / 2
ax.annotate(
"",
xy=(1.3, E_H2_theo),
xytext=(1.3, 2 * E_H_theo),
arrowprops=dict(arrowstyle="<->", color="g"),
)
ax.text(
1.35, mid_y_theo, f"ΔE: {E_reaction_theo:.4f}", color="g", verticalalignment="top"
)
ax.annotate(
"",
xy=(1.5, E_H2_exp),
xytext=(1.5, 2 * E_H_exp),
arrowprops=dict(arrowstyle="<->", color="g", linestyle="dashed"),
)
ax.text(
1.55,
mid_y_exp,
f"ΔE_VQE: {E_reaction_exp:.4f}",
color="g",
verticalalignment="center",
)
# Add legend
ax.legend()
plt.show()

Come mostrato nella figura, sebbene ci siano alcuni errori, l'energia dello stato fondamentale esatta degli Hamiltoniani e l'energia di reazione calcolata usando i risultati VQE sono simili, vicine a -0,2 Hartree.
Va notato che l'energia di reazione di questo processo ha un valore negativo, il che significa che l'energia viene rilasciata attraverso il processo, e la molecola risultante ha un'energia inferiore rispetto a due atomi singoli. Riassumiamo ciò che abbiamo imparato finora.
Abbiamo prima esaminato due importanti tecniche di approssimazione necessarie per risolvere i problemi di chimica quantistica: il principio variazionale e la scelta del set di base, entrambi fondamentali per VQE. Abbiamo esplorato il principio variazionale manualmente, calcolando l'energia dello stato fondamentale dell'oscillatore armonico semplice.
Successivamente, abbiamo esplorato VQE, un algoritmo ampiamente utilizzato per calcolare l'energia dello stato fondamentale di un sistema quantistico. Abbiamo eseguito del codice per calcolare le energie dello stato fondamentale dell'idrogeno atomico () e della molecola di idrogeno (). In particolare, abbiamo imparato che è necessario ottenere l'Hamiltoniano molecolare appropriato per il sistema e trasformarlo in una forma eseguibile su un computer quantistico. Abbiamo anche visto che l'ansatz, un circuito quantistico parametrizzato, è necessario per preparare stati quantistici di prova all'interno di VQE, e abbiamo discusso l'importanza di scegliere un'adeguata struttura del circuito ansatz. Abbiamo inoltre imparato che VQE si basa su un processo di ottimizzazione iterativa che utilizza un computer classico, guidando il circuito quantistico a trovare lo stato di energia minima, e abbiamo visto come il processo converge.
Infine, abbiamo usato le energie dello stato fondamentale calcolate di e ottenute tramite VQE per calcolare l'energia di reazione per il processo .
VQE è un potente algoritmo quantistico a breve termine, ma è importante essere consapevoli dei suoi limiti. Le prestazioni di VQE dipendono fortemente dalla scelta dell'ansatz — trovare un ansatz efficiente da preparare che possa rappresentare accuratamente il vero stato fondamentale diventa difficile per molecole più grandi e complesse. Inoltre, l'hardware quantistico attuale è suscettibile al rumore, che può influire sulla precisione dei risultati VQE, in particolare per circuiti più profondi o con un numero maggiore di qubit. Nonostante queste sfide, VQE funge da algoritmo fondamentale, e la ricerca in corso sta esplorando metodi variazionali più sofisticati e tecniche di mitigazione degli errori per ampliare i limiti di ciò che è possibile nella chimica quantistica su computer quantistici a breve termine. Ad esempio, algoritmi come la Diagonalizzazione Quantistica Basata su Campioni (SQD) sono in fase di sviluppo, sfruttando campioni ottenuti da circuiti quantistici combinati con la diagonalizzazione classica in un sottospazio per migliorare la stima dell'energia e affrontare alcune delle limitazioni di VQE, in particolare riguardo all'efficienza delle misure e alla robustezza al rumore.
Revisione e domande
Concetti fondamentali:
- L'algoritmo variazionale quantistico è un paradigma di calcolo in cui un computer classico e un computer quantistico lavorano insieme per risolvere un problema.
- In VQE, si parte dall'Hamiltoniano del sistema e lo si mappa sui qubit per l'esecuzione sul computer quantistico. Si seleziona un circuito quantistico parametrizzato, un ansatz, e si eseguono misure ripetute variando i parametri dell'ansatz, fino a raggiungere il valore energetico minimo. La ricerca nello spazio dei parametri viene eseguita utilizzando un ottimizzatore classico. Per ottenere buoni risultati, è necessario selezionare un buon ansatz e un ottimizzatore appropriato.
- L'energia di reazione è la variazione totale di energia in una reazione chimica, determinata dalla differenza tra l'energia dei reagenti e dei prodotti.
Vero/falso
- Il principio variazionale afferma che il valore di aspettazione dell'energia per qualsiasi funzione d'onda di prova è sempre maggiore o uguale alla vera energia dello stato fondamentale.
- Un set di base è una raccolta di funzioni usate per approssimare le funzioni d'onda quantistiche.
- VQE è un algoritmo quantistico usato per risolvere esattamente l'equazione di Schrödinger per un dato Hamiltoniano.
- In VQE, un circuito quantistico parametrizzato (un ansatz) viene usato per preparare funzioni d'onda di prova.
- La scelta dell'ottimizzatore in VQE (ad esempio, COBYLA, SPSA o ADAM) non influisce sulla qualità del risultato.
- L'
Estimatordi Qiskit è usato per calcolare direttamente i valori di aspettazione degli Hamiltoniani in VQE.
Domande a scelta multipla:
- Qual è lo scopo dell'Hamiltoniano in VQE?
- A) Generare stati quantistici casuali
- B) Determinare l'energia degli stati quantistici
- C) Ottimizzare i circuiti quantistici
- D) Creare entanglement
- Qual è l'obiettivo principale dell'algoritmo VQE?
- A) Trovare l'energia dello stato fondamentale di un Hamiltoniano
- B) Creare entanglement tra i qubit
- C) Eseguire la ricerca di Grover
- D) Rompere la cifratura RSA
- Quanti stati quantistici vengono generati in questo notebook per confrontare l'ansatz?
- A) 100
- B) 1000
- C) 5000
- D) 10.000
- Perché è necessario un ottimizzatore classico in VQE?
- A) Per eseguire le misure quantistiche
- B) Per aggiornare i parametri dell'ansatz e minimizzare l'energia
- C) Per creare entanglement tra i qubit
- D) Per generare casualità quantistica
- Perché l'ansatz è progettato per essere parametrizzato?
- A) Per consentire la preparazione degli stati quantistici
- B) Per permettere la ricerca in un ampio spazio di stati quantistici
- C) Per ridurre la complessità del circuito
- D) Per misurare direttamente gli autovalori
- Quale delle seguenti è l'affermazione più corretta riguardo alla scelta di un buon ansatz?
- A) Un ansatz deve produrre stati distribuiti uniformemente sulla sfera di Bloch, altrimenti fallirà.
- B) Un ansatz dovrebbe essere adattato al tuo sistema per assicurarsi che possa generare stati vicini allo stato fondamentale.
- C) Un ansatz dovrebbe produrre stati casuali usando i suoi parametri variazionali.
- D) Un ansatz migliore ha sempre più parametri variazionali.
(Opzionale) Appendice: Overhead dell'ottimizzatore in funzione della complessità dell'ansatz
VQE affronta diverse sfide ben note [ref 6], e le seguenti sono correlate a ciò che abbiamo imparato sopra.
- Sfide nella selezione dell'ansatz
Esiste una sfida intrinseca nella scelta del giusto ansatz variazionale. Gli ansätze ispirati alla chimica (come UCCSD) offrono accuratezza fisica ma richiedono circuiti profondi, mentre gli ansätze efficienti per l'hardware hanno circuiti più superficiali ma possono mancare di interpretabilità fisica. Inoltre, molti ansätze introducono parametri variazionali eccessivi che contribuiscono poco a migliorare la precisione ma aumentano significativamente la difficoltà di ottimizzazione.
- Difficoltà di ottimizzazione
Il panorama di ottimizzazione di VQE può avere regioni in cui i gradienti svaniscono esponenzialmente (altopiani sterili), rendendo difficile per gli ottimizzatori classici aggiornare i parametri variazionali in modo efficiente. Per questo, i ricercatori hanno tentato di usare diversi tipi di ottimizzatori — basati su gradiente e senza gradiente — ma entrambi affrontano delle sfide. Gli ottimizzatori basati sul gradiente soffrono degli altopiani sterili, mentre i metodi senza gradiente richiedono un gran numero di valutazioni della funzione.
- Overhead dell'ottimizzatore
Un'altra sfida ben nota è l'overhead dell'ottimizzatore, che è legato alla scala del problema. I circuiti quantistici richiesti da VQE crescono in profondità e complessità all'aumentare della dimensione del problema; questo di solito aumenta anche il numero di parametri da ottimizzare. Il processo di ottimizzazione diventa intrattabile all'aumentare del numero di parametri, portando a una convergenza lenta e a difficoltà nel trovare la soluzione ottimale.
Qui esamineremo queste sfide usando VQE per una molecola , con due diversi tipi di ansätze.
(Nota: Questo può richiedere più tempo QPU, quindi sentiti libero di usare un simulatore se non hai abbastanza tempo.)
from qiskit.circuit import ParameterVector
num_iter = 4
alpha = ParameterVector("alpha", 3)
beta = ParameterVector("beta", 3 * num_iter)
# step1: Map problem to quantum circuits and operators
hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
ansatz_1 = ansatz3
ansatz_2 = QuantumCircuit(1)
for i in range(num_iter):
ansatz_2.rx(beta[i * 3 + 0], 0)
ansatz_2.rz(beta[i * 3 + 1], 0)
ansatz_2.rx(beta[i * 3 + 2], 0)
ansatz_1.draw("mpl")
ansatz_2.draw("mpl")

# Step 2: Optimize for target hardware
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa_1 = pm.run(ansatz_1)
ansatz_isa_2 = pm.run(ansatz_2)
hamiltonian_isa_1 = hamiltonian.apply_layout(layout=ansatz_isa_1.layout)
hamiltonian_isa_2 = hamiltonian.apply_layout(layout=ansatz_isa_2.layout)
Ora eseguiamo un VQE con un punto iniziale composto da tutti uno, con un massimo di 20 passi, e confrontiamo la convergenza di entrambe le esecuzioni.
# QPU time 3m 40s for ibm_brisbane
# Step 3: Execute on target hardware
from scipy.optimize import minimize
x0 = np.ones(ansatz_1.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_1, hamiltonian_isa_1, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.8782202668652658]
Iters. done: 2 [Current cost: -0.43473160695469165]
Iters. done: 3 [Current cost: -0.4076372093159749]
Iters. done: 4 [Current cost: -1.3587839859772106]
Iters. done: 5 [Current cost: -1.774529906754082]
Iters. done: 6 [Current cost: -1.541934983115727]
Iters. done: 7 [Current cost: -1.2732403113465345]
Iters. done: 8 [Current cost: -1.820842221085785]
Iters. done: 9 [Current cost: -1.8065762857059005]
Iters. done: 10 [Current cost: -1.8126394095981146]
Iters. done: 11 [Current cost: -1.8205831886180421]
Iters. done: 12 [Current cost: -1.8086715778994924]
Iters. done: 13 [Current cost: -1.8307676638629322]
Iters. done: 14 [Current cost: -1.8177328827556327]
Iters. done: 15 [Current cost: -1.8179426218088064]
Iters. done: 16 [Current cost: -1.8109239667991088]
Iters. done: 17 [Current cost: -1.824271872489647]
Iters. done: 18 [Current cost: -1.813167587671394]
Iters. done: 19 [Current cost: -1.824647343397313]
Iters. done: 20 [Current cost: -1.8219785311686143]
# Save Cost_history as a new list
ansatz_1_history = cost_history_dict["cost_history"]
# QPU time 3m 40s for ibm_brisbane
x0 = np.ones(ansatz_2.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_2, hamiltonian_isa_2, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.738191173881188]
Iters. done: 2 [Current cost: -0.42636037194506304]
Iters. done: 3 [Current cost: -1.3503788613797374]
Iters. done: 4 [Current cost: -0.9109204349776897]
Iters. done: 5 [Current cost: -0.9060873157510835]
Iters. done: 6 [Current cost: -0.7735065414083984]
Iters. done: 7 [Current cost: -1.586889197437709]
Iters. done: 8 [Current cost: -1.659215191584943]
Iters. done: 9 [Current cost: -1.245445981794618]
Iters. done: 10 [Current cost: -1.1608385766138023]
Iters. done: 11 [Current cost: -1.1551733876027737]
Iters. done: 12 [Current cost: -1.8143337768286332]
Iters. done: 13 [Current cost: -1.2510951563756598]
Iters. done: 14 [Current cost: -1.6918311531865413]
Iters. done: 15 [Current cost: -1.8163783305531838]
Iters. done: 16 [Current cost: -1.8434877732947152]
Iters. done: 17 [Current cost: -1.8461898233304472]
Iters. done: 18 [Current cost: -1.0346471214915485]
Iters. done: 19 [Current cost: -1.8322518854150687]
Iters. done: 20 [Current cost: -1.717144678705999]
ansatz_2_history = cost_history_dict["cost_history"]
fig, ax = plt.subplots()
# Define the constant function)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_1_history,
label="Ansatz with 3 parameters",
)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_2_history,
label="Ansatz with 12 parameters",
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
plt.legend()
plt.draw()

Il grafico sopra dimostra chiaramente che il processo di ottimizzazione dell'ansatz con più variabili richiede più tempo per raggiungere una convergenza stabile.
Piuttosto che affidarsi a semplici circuiti a singolo qubit e un ansatz diretto, la complessità dell'ottimizzazione aumenta quando sono richiesti circuiti quantistici più grandi e ansätze con strutture più complesse. Questo evidenzia una sfida ben nota nei VQE: l'overhead dell'ottimizzatore.
I ricercatori continuano a sviluppare varie metodologie avanzate che possono utilizzare i computer quantistici per i problemi di chimica. Puoi accedere a una varietà di materiali didattici su IBM Quantum Learning.
Riferimenti
- [ref 1 ] Richard P. Feynman, Simulating Physics with Computers, International Journal of Theoretical Physics, 1982.
- [ref 2] Marov, M.Y. (2015). The Structure of the Universe. In: The Fundamentals of Modern Astrophysics. Springer, New York, NY.
- [ref 3] How to solve difficult chemical engineering problems with quantum computing, IBM Research Blog, 2023.
- [ref 4] Y. Cao, J. Romero and A. Aspuru-Guzik, "Potential of quantum computing for drug discovery," in IBM Journal of Research and Development, vol. 62, no. 6, pp. 6:1-6:20, 1 Nov.-Dec. 2018
- [ref 5] Present State of Molecular Structure Calculation, REv. Mod. Phys. 32, 170, 1960
- [ref 6] Fedorov, D.A., Peng, B., Govind, N. et al. VQE method: a short survey and recent developments. Mater Theory 6, 2 (2022)