HI-VQE Chemistry - Una Qiskit Function di Qunova Computing
# Added by doQumentation — required packages for this notebook
!pip install -q qiskit-ibm-catalog qiskit-ibm-runtime
# This cell is hidden from users
from qiskit_ibm_runtime import QiskitRuntimeService
service = QiskitRuntimeService()
instance = service.active_account()["instance"]
backend_name = service.least_busy(operational=True, min_num_qubits=16).name
qiskit-ibm-runtime~=0.45.0
Le Qiskit Functions sono una funzionalità sperimentale disponibile esclusivamente per gli utenti dei piani IBM Quantum® Premium, Flex e On-Prem (tramite IBM Quantum Platform API). Sono in stato di anteprima e soggette a modifiche.
Panoramica
Nella chimica quantistica, il problema della struttura elettronica consiste nel trovare le soluzioni all'equazione di Schrödinger elettronica — le funzioni d'onda quantistiche che descrivono il comportamento degli elettroni del sistema. Queste funzioni d'onda sono vettori di ampiezze complesse, ciascuna corrispondente al contributo di una possibile configurazione elettronica.
Lo stato fondamentale è la funzione d'onda del sistema a energia minima e riveste un'importanza particolare nello studio dei sistemi molecolari. L'approccio più accurato per calcolare lo stato fondamentale considera tutte le possibili configurazioni elettroniche, ma questo diventa intrattabile per sistemi più grandi, poiché il numero di configurazioni cresce esponenzialmente con la dimensione del sistema.
L'Handover Iterative Variational Quantum Eigensolver (HI-VQE) è un innovativo metodo ibrido quantistico-classico per stimare con precisione lo stato fondamentale di sistemi molecolari. Integra hardware quantistico con elaborazione classica, usando processori quantistici per esplorare efficientemente le configurazioni elettroniche candidate e calcolando la funzione d'onda risultante su computer classici. Generando funzioni d'onda compatte ma chimicamente accurate, HI-VQE potenzia la ricerca e la scoperta nella chimica quantistica e nella scienza dei materiali.

HI-VQE riduce la complessità computazionale del problema della struttura elettronica stimando efficientemente lo stato fondamentale con elevata precisione. Si concentra su un sottoinsieme accuratamente selezionato delle configurazioni elettroniche più rilevanti, ottimizzando sia l'accuratezza che l'efficienza.
Combinando i punti di forza dei computer classici e quantistici, HI-VQE affina e migliora iterativamente la stima attuale della funzione d'onda. Le sue tecniche uniche di costruzione del sottospazio contribuiscono a rendere la selezione delle configurazioni più efficiente, offrendo agli utenti un maggiore controllo computazionale e una precisione migliorata nelle simulazioni di chimica quantistica.
Se desideri approfondire l'algoritmo, puoi leggere il relativo articolo di ricerca.
Descrizione
Il numero di configurazioni elettroniche per un sistema molecolare cresce esponenzialmente con la dimensione del sistema. Tuttavia, per certi stati elettronici, come lo stato fondamentale, è comune che solo una piccola frazione delle configurazioni contribuisca significativamente all'energia dello stato. I metodi Selected Configuration Interaction (SCI) sfruttano questa sparsità per ridurre i costi computazionali, identificando e concentrandosi sulle configurazioni più rilevanti. Questo sottoinsieme di configurazioni è detto sottospazio.
HI-VQE sfrutta l'efficienza intrinseca dei computer quantistici nella rappresentazione dei sistemi molecolari per supportare la ricerca del sottospazio. Integra subroutine classiche e quantistiche per risolvere il problema della struttura elettronica con elevata precisione. A differenza dei metodi SCI quantistici esistenti, HI-VQE combina addestramento variazionale, costruzione iterativa del sottospazio e screening delle configurazioni tramite pre-diagonalizzazione per migliorare l'efficienza riducendo le misurazioni quantistiche, le iterazioni e i costi di diagonalizzazione classica. HI-VQE può quindi essere applicato a sistemi molecolari più grandi che richiedono più qubit, e riduce il costo per risolvere un problema di una data dimensione allo stesso grado di accuratezza.
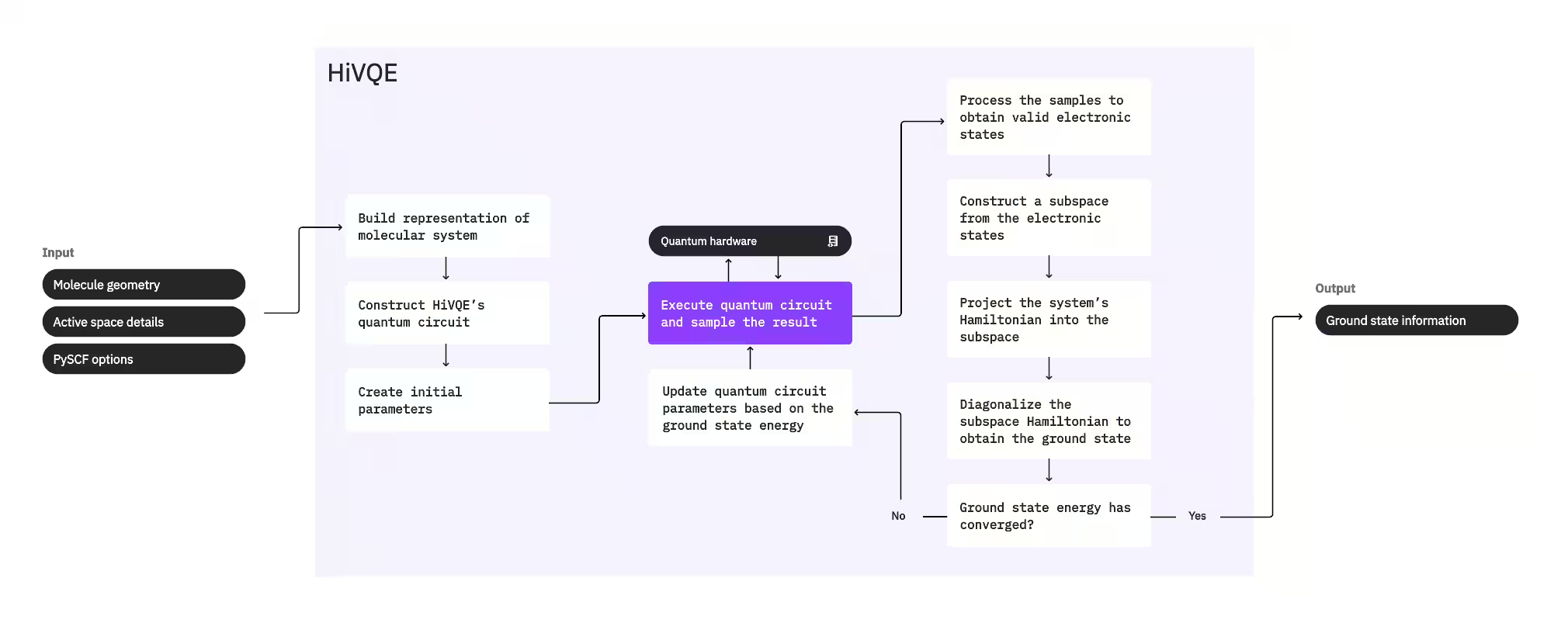
Per calcolare lo stato fondamentale di un sistema, HI-VQE utilizza dapprima il pacchetto di chimica classica PySCF per generare una rappresentazione molecolare a partire dagli input forniti dall'utente, come la geometria molecolare e altre informazioni sul sistema. Entra quindi in un ciclo di ottimizzazione ibrido quantistico-classico, raffinando iterativamente un sottospazio per rappresentare ottimalmente lo stato fondamentale riducendo al minimo il numero di configurazioni incluse. Il ciclo continua fino al soddisfacimento dei criteri di convergenza, come la dimensione del sottospazio o la stabilità dell'energia, dopodiché vengono restituiti la funzione d'onda dello stato fondamentale calcolata e la sua energia. Questi risultati possono essere utilizzati per costruire superfici di energia potenziale accurate ed eseguire ulteriori analisi del sistema.
Il ciclo di ottimizzazione si concentra sull'adeguamento dei parametri di un circuito quantistico per generare un sottospazio di alta qualità. HI-VQE offre tre opzioni di circuito quantistico: excitation_preserving, efficient_su2 e LUCJ. L'ottimizzazione viene inizializzata in prossimità dello stato di riferimento Hartree-Fock per la sua idoneità generale. Il circuito viene poi eseguito su un dispositivo quantistico e le configurazioni vengono campionate dallo stato quantistico risultante, prima di essere restituite come stringhe binarie. A causa del rumore del dispositivo quantistico, alcune configurazioni campionate potrebbero essere fisicamente non valide, non conservando il numero di elettroni o lo spin. HI-VQE risolve questo problema usando il processo di configuration recovery del pacchetto qiskit-addon-sqd, in modo che gli utenti possano correggere le configurazioni non valide o scartarle.
Le configurazioni valide subiscono poi un passaggio di screening opzionale per rimuovere quelle che si prevede contribuiscano in misura minima. Questo riduce la dimensione del sottospazio, abbassando così il costo del passaggio di diagonalizzazione. Se lo screening è abilitato, viene costruita un'Hamiltoniana del sottospazio preliminare dalle configurazioni valide e viene eseguita una diagonalizzazione con criteri di terminazione molto approssimati. Sebbene la precisione delle ampiezze risultanti per ciascuna configurazione sia bassa, questo metodo è efficace per prevedere quali configurazioni escludere dal sottospazio in quella iterazione ed è rapido da calcolare.
Le configurazioni selezionate vengono aggiunte al sottospazio e l'Hamiltoniana del sistema viene proiettata in questo sottospazio. Il sottospazio si aggiorna iterativamente, preservando le configurazioni più rilevanti attraverso le iterazioni. Questo approccio si distingue dai metodi alternativi perché il circuito quantistico non deve approssimare lo stato fondamentale completo ad ogni passo.
Successivamente, l'Hamiltoniana del sottospazio viene diagonalizzata classicamente per ottenere il valore proprio minimo e il corrispondente autovettore, che rappresentano un'approssimazione dello stato fondamentale e della sua energia. Con il miglioramento della qualità del sottospazio attraverso le iterazioni, lo stato fondamentale calcolato approssima sempre meglio il vero stato fondamentale. A questo punto può essere eseguito un ulteriore passaggio di screening per rimuovere dal sottospazio le configurazioni che non contribuiscono in modo sostanziale allo stato fondamentale calcolato. Questo passo garantisce che il sottospazio portato nell'iterazione successiva sia il più compatto possibile. La valutazione si basa sulle ampiezze restituite dalla diagonalizzazione, poiché queste rappresentano il contributo di importanza di ciascuna configurazione allo stato fondamentale calcolato.
Un controllo di convergenza determina quindi se ulteriori iterazioni migliorerebbero i risultati. In caso affermativo, viene eseguito un opzionale passo di espansione classica, i parametri del circuito quantistico vengono aggiornati per minimizzare ulteriormente l'energia calcolata e il processo si ripete. Il passo di espansione classica genera configurazioni aggiuntive per il sottospazio, integrando quelle campionate dal dispositivo quantistico. Individua prima la configurazione con l'ampiezza maggiore nei risultati della diagonalizzazione, per poi generare nuove configurazioni con singole e doppie eccitazioni a partire dalla configurazione identificata. Il numero desiderato di queste configurazioni viene poi aggiunto al sottospazio.
Una volta stabilito che le iterazioni hanno raggiunto la convergenza, HI-VQE restituisce lo stato fondamentale calcolato (sotto forma degli stati nel sottospazio e delle loro ampiezze nella funzione d'onda dello stato fondamentale), la sua energia e una misura della varianza dell'energia che indica se lo stato calcolato costituisce un autostato dell'Hamiltoniana del sistema.
Gli utenti possono scegliere il circuito quantistico utilizzato e il numero di shot per ciascun circuito quantistico, nonché controllare la dimensione del sottospazio o abilitare la generazione classica di configurazioni aggiuntive a supporto di quelle generate quantisticamente. In questo modo, gli utenti possono adattare il comportamento di HI-VQE alle loro applicazioni specifiche.
Licenza
Si prega di notare che l'utilizzo di questa Qiskit Function è limitato ai problemi che richiedono al massimo 20 qubit, salvo l'ottenimento di una licenza che conceda un limite superiore.
Si prega di inviare un'e-mail a qiskit.support@qunovacomputing.com per informazioni sull'ottenimento di una licenza.
Per iniziare
Prima di tutto, richiedi l'accesso alla funzione. Poi, autenticati con la tua chiave API IBM Quantum® e, avendo già salvato il tuo account nell'ambiente locale, seleziona la Qiskit Function come segue:
import reprlib
from qiskit_ibm_catalog import QiskitFunctionsCatalog
catalog = QiskitFunctionsCatalog(channel="ibm_quantum_platform")
function = catalog.load("qunova/hivqe-chemistry")
Esempio
Il primo esempio mostra come calcolare l'energia dello stato fondamentale per una molecola di NH3 usando l'algoritmo HI-VQE.
Definire la geometria molecolare e le opzioni
La geometria molecolare di NH3 viene fornita con coordinate cartesiane separate da ";" per ciascun atomo.
# Define the molecule geometry
geometry = """
N -0.85188 -0.02741 0.03141;
H 0.16545 0.00593 -0.01648;
H -1.16348 -0.39357 -0.86702;
H -1.16348 0.94228 0.06281;
"""
È possibile definire e fornire opzioni aggiuntive per il sistema molecolare nel seguente formato dizionario.
# Configure some options for the job.
molecule_options = {"basis": "sto3g"}
hivqe_options = {"shots": 100, "max_iter": 20}
Esegui la funzione con la geometria e le opzioni come input.
# Run HI-VQE
job = function.run(
geometry=geometry,
# `backend_name` is the name of a backend with at least 16 qubits,
# for example, "ibm_marrakesh".
backend_name=backend_name,
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
È buona pratica stampare l'ID del job della Function in modo da poterlo fornire nelle richieste di supporto in caso di problemi.
print("Job ID:", job.job_id)
Job ID: e5ced6f2-fd1d-4244-a6aa-bd27cfb0cdee
Questo esempio utilizza 16 qubit con 8 orbitali di base sto3g per una molecola di NH3. Controlla lo stato del workload della tua Qiskit Function o recupera i risultati come segue:
print(job.status())
QUEUED
Una volta completato il job, i risultati possono essere ottenuti con l'istanza result().
result = job.result()
# Output can be long, so we display a shortened representation
shortened_result = reprlib.repr(result)
print(shortened_result)
{'eigenvector': [0.9824448589364075, 0.009527106392132133, 6.854074372058527e-08, 3.591500190038039e-07, 0.0012975231577544268, 2.310159709002111e-05, ...], 'energy': -55.52108557170985, 'energy_history': [-55.51901898989887, -55.52056881448526, -55.52065046778772, -55.520690696813716, -55.520691108428, -55.520708448092634, ...], 'energy_variance': 3.066239097617371e-10, ...}
Per accedere all'energia dello stato fondamentale, usa la chiave "energy". La chiave "eigenvector" fornisce i coefficienti CI con la corrispondente notazione a stringa di bit della configurazione elettronica memorizzata in "states" dei risultati.
fci_energy = -55.521148034704126 # the exact energy using FCI method
hivqe_energy = result["energy"]
print(
f"|Exact Energy - HI-VQE Energy|: "
f"{abs(fci_energy - hivqe_energy) * 1000} mHa"
)
print(f"Sampled Number of States: {len(result['states'])}")
|Exact Energy - HI-VQE Energy|: 0.06246299427914437 mHa
Sampled Number of States: 1936
Prestazioni
Questa sezione mostra i calcoli di benchmark dimostrati di HI-VQE con un caso a 24 qubit per Li2S, un caso a 40 qubit per una molecola di N2 e un caso a 44 qubit per un sistema FeP-NO.
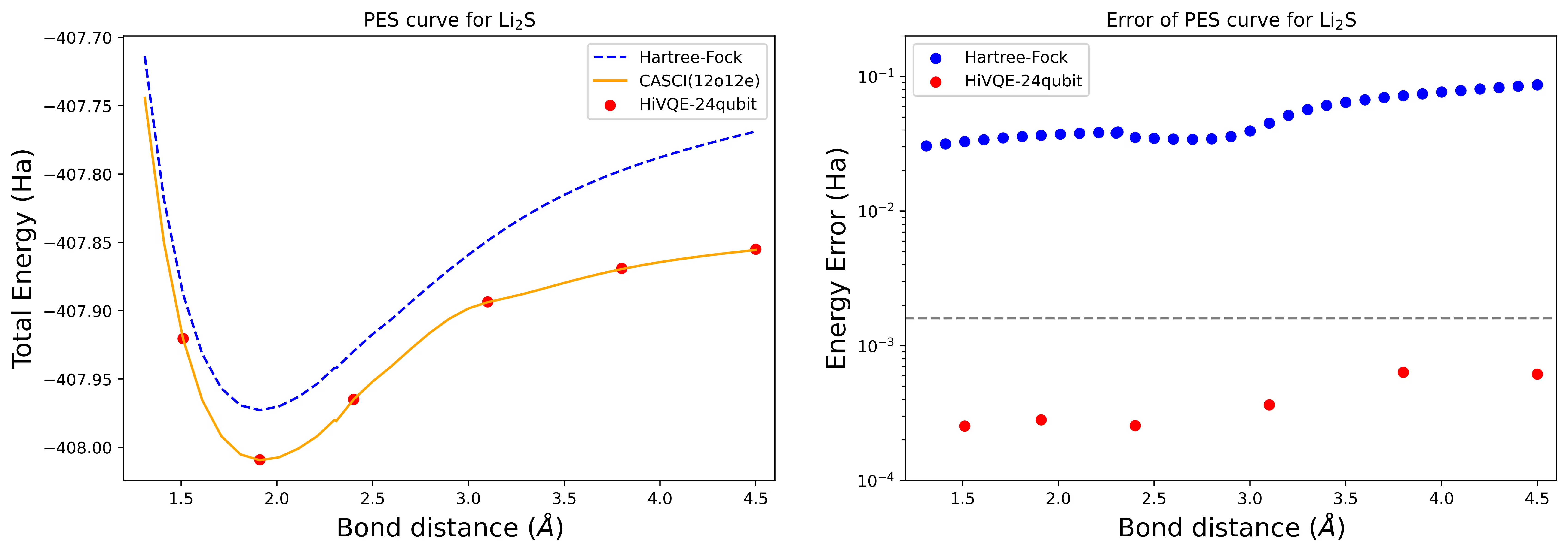
Curva della superficie di energia potenziale di dissociazione per una molecola di Li2S con 24 qubit
La curva PES è mostrata con il riferimento FCI e la stima iniziale da RHF, insieme all'errore di energia rispetto al riferimento FCI.
I calcoli sono stati condotti con le seguenti geometrie e opzioni.
# This cell is hidden from users
backend_name = service.least_busy(operational=True, min_num_qubits=38).name
# Define Li2S geometries
Li2S_geoms = {
"Li2S_1.51": "S -1.239044 0.671232 -0.030374;Li -1.506327 0.432403 -1.498949;Li -0.899996 0.973348 1.826768;",
"Li2S_2.40": "S -1.741432 0.680397 0.346702;Li -0.529307 0.488006 -1.729343;Li -1.284307 0.989409 2.177209;",
"Li2S_3.80": "S -2.707255 0.674298 0.909161;Li 0.079218 0.552012 -1.671656;Li -0.927010 0.931502 1.557063;",
}
# Configure some options for the job.
molecule_options = {
"basis": "sto3g",
}
hivqe_options = {
"shots": 100,
"max_iter": 20,
}
results = []
for geom in ["Li2S_1.51", "Li2S_2.40", "Li2S_3.80"]:
# Run HI-VQE
job = function.run(
geometry=Li2S_geoms[geom],
backend_name=backend_name, # can use any device with at least 38 qubits
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
results.append(job.result())
I punti rossi rappresentano i risultati del calcolo HI-VQE per sei geometrie diverse; tre geometrie corrispondenti a 1,51, 2,40 e 3,80 Angstrom sono fornite come input nella cella precedente.
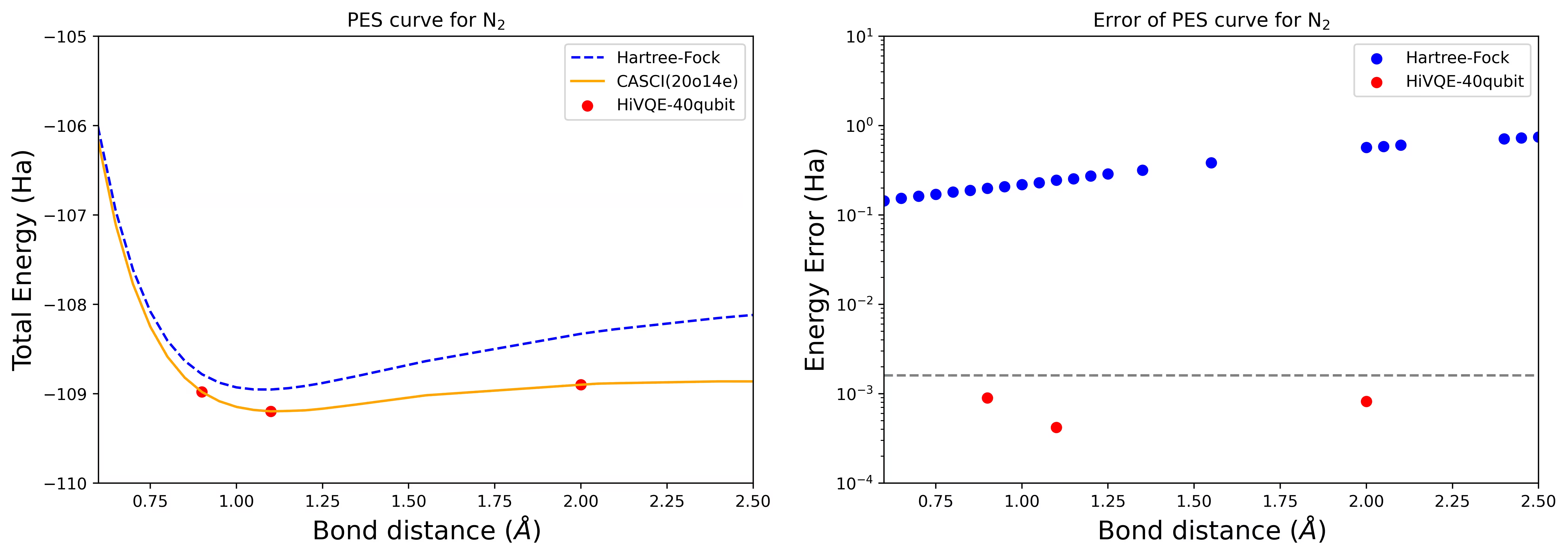
Curva PES di dissociazione per una molecola di N2 con 40 qubit
La molecola di azoto è stata identificata come un sistema multiriferimento con grandi contributi di energia di correlazione oltre lo stato Hartree-Fock. Abbiamo condotto un calcolo di benchmark per la molecola di N2 con base cc-pvdz, (20o,14e) usando la selezione degli orbitali attivi homo-lumo. Il numero di spazio attivo completo (CAS) per rappresentare questo problema è 6.009.350.400. Non è possibile ottenere la soluzione del problema agli autovalori (per energia e struttura elettronica) con questo numero di stati usando un potente desktop (16cpu/64GB). Con HI-VQE, gli utenti possono cercare efficientemente il sottospazio degli stati CAS per trovare risultati chimicamente accurati, riducendo significativamente le risorse computazionali. I grafici seguenti mostrano la curva PES del calcolo HI-VQE a 40 qubit della dissociazione della molecola di N2.
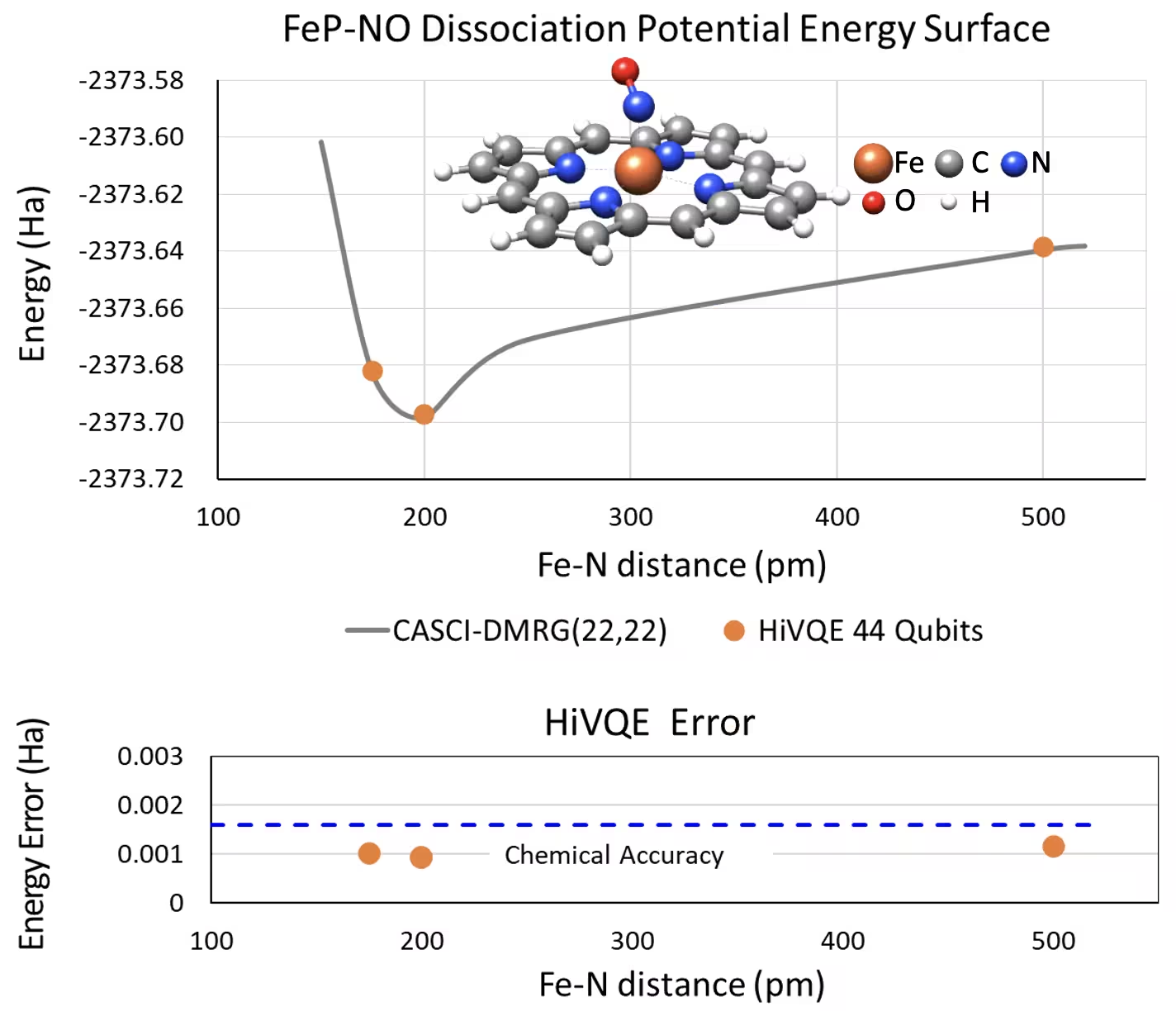
Curva PES di dissociazione per il ferro(II)-porfirina pentacoordinata con un sistema NO a 44 qubit
Un altro sistema chimico interessante è un complesso di ferro(II)-porfirina (FeP) con un legante ossido nitrico (NO) coordinato, che rappresenta un sistema metalloporfirina biologicamente rilevante con ruoli cruciali in vari processi fisiologici. In questo esempio, HI-VQE è stato utilizzato per stimare la curva accurata della superficie di energia potenziale dell'interazione intermolecolare tra FeP e NO (energia dello stato fondamentale per geometrie a diversa separazione). Il sistema combinato ha 450 orbitali e 202 elettroni (450o,202e) con base 6-31g(d) in totale. La selezione degli orbitali attivi homo-lumo è stata utilizzata per calcolare il caso più piccolo rispetto al caso reale con (22o,22e). Dai seguenti risultati di benchmark, siamo stati in grado di raggiungere la precisione chimica (> 1,6 mHa) con un calcolo di chimica su computer classico all'avanguardia di CASCI(DMRG) (22o,22e) come riferimento.
Benchmark
- La dimensione esatta della matrice è il numero di determinanti per la soluzione esatta, come FCI e CASCI.
- Il calcolo HI-VQE campiona e calcola il sottospazio di essa (cioè la dimensione della matrice HI-VQE).
- Il tempo totale include il tempo di esecuzione su QPU e le esecuzioni della Qiskit Function su CPU.
- La precisione è stimata dalla differenza di energia rispetto alla soluzione esatta.
| Sistema chimico | Numero di qubit | Dimensione esatta della matrice | Dimensione matrice HI-VQE | E(diff) dall'esatto (mHa) | Numero di iterazioni | Tempo totale | Utilizzo tempo QPU |
|---|---|---|---|---|---|---|---|
| (8o,10e) | 16 | 3136 | 1936 | 0,08 | 6 | 37 s | 34 s |
| (10o,10e) | 20 | 63504 | 3969 | 0,60 | 5 | 250 s | 50 s |
| (15o,10e) | 30 | 9018009 | 49729 | 0,90 | 5 | 354 s | 54 s |
| (16o,14e) | 32 | 130873600 | 1798281 | 1,10 | 9 | 6531 s | 121 s |
| (18o,24e) | 36 | 344622096 | 399424 | 0,90 | 24 | 5174 s | 130 s |
| (20o,14e) | 40 | 6009350400 | 9012004 | 1,20 | 21 | 46547 s | 258 s |
Recuperare i messaggi di errore
Se il tuo workload fallisce, lo stato sarà ERROR e la chiamata a job.result() solleverà un'eccezione:
job = function.run(
geometry="invalid-geometry", # This will cause an error
backend_name=backend_name,
max_states=2000,
max_expansion_states=15,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
job.result()
job.status()
'ERROR'
Ottenere supporto
Puoi inviare un'e-mail a qiskit.support@qunovacomputing.com per ricevere assistenza con questa funzione.
Se hai bisogno di aiuto per risolvere un errore specifico, fornisci l'ID del job della Function che ha riscontrato l'errore.
Passi successivi
- Richiedi l'accesso alla funzione compilando questo modulo.
- Visita il riferimento API per questa Qiskit Function.
- Prova il tutorial Compute dissociation PES curve for FeP-NO with HI-VQE.
- Consulta Pellow-Jarman, A., et al. (2025). HIVQE: Handover Iterative Variational Quantum Eigensolver for Efficient Quantum Chemistry Calculations. arXiv preprint arXiv:2503.06292.
- Prova il tutorial Dissociation PES curves with Qunova HiVQE.