Migliorare la stima dell'energia di un Hamiltoniano chimico con SQD
In questo tutorial implementiamo un pattern Qiskit che mostra come post-elaborare campioni quantistici rumorosi per trovare un'approssimazione allo stato fondamentale di un Hamiltoniano chimico: la molecola all'equilibrio nel set di basi 6-31G. Seguiremo un approccio di diagonalizzazione quantistica basata su campioni per elaborare campioni prelevati da un ansatz di circuito quantistico a 36 qubit (in questo caso, un circuito LUCJ). Per tenere conto dell'effetto del rumore quantistico, viene utilizzata la tecnica di recupero della configurazione.
Il pattern può essere descritto in quattro passaggi:
- Passaggio 1: Mappatura al problema quantistico
- Generare un ansatz per stimare lo stato fondamentale
- Passaggio 2: Ottimizzare il problema
- Transpilare l'ansatz per il backend
- Passaggio 3: Eseguire gli esperimenti
- Estrarre campioni dall'ansatz usando la primitiva
Sampler
- Estrarre campioni dall'ansatz usando la primitiva
- Passaggio 4: Post-elaborare i risultati
- Ciclo di recupero della configurazione auto-consistente
- Post-elaborare l'insieme completo di campioni di bitstring, usando la conoscenza a priori del numero di particelle e l'occupazione orbitale media calcolata all'iterazione più recente.
- Creare probabilisticamente batch di sottocampioni da bitstring recuperate.
- Proiettare e diagonalizzare l'Hamiltoniano molecolare su ciascun sottospazio campionato.
- Salvare l'energia dello stato fondamentale minima trovata in tutti i batch e aggiornare l'occupazione orbitale media.
- Ciclo di recupero della configurazione auto-consistente
Per questo esempio, l'Hamiltoniano degli elettroni interagenti assume la forma generica:
/ sono gli operatori fermionic di creazione/annichilazione associati al -esimo elemento del set di basi e allo spin . e sono gli integrali elettronici a uno e due corpi.
Il flusso di lavoro SQD con recupero della configurazione auto-consistente è illustrato nel seguente diagramma.
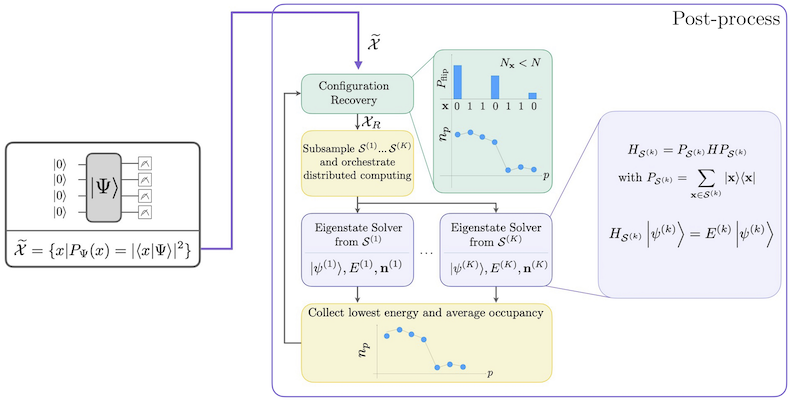
È noto che SQD funziona bene quando l'autostato target è sparso: la funzione d'onda è supportata in un insieme di stati di base la cui dimensione non cresce esponenzialmente con la dimensione del problema. In questo scenario, la diagonalizzazione dell'Hamiltoniano proiettato nel sottospazio definito da :
produce una buona approssimazione all'autostato target. Il ruolo del dispositivo quantistico è quello di produrre campioni dei membri di soltanto. Prima, un circuito quantistico prepara lo stato nel dispositivo quantistico. Viene usata la codifica Jordan-Wigner. Di conseguenza, i membri della base computazionale rappresentano stati di Fock (configurazioni/determinanti elettronici). Il circuito viene campionato nella base computazionale, producendo l'insieme di configurazioni rumorose . Le configurazioni sono rappresentate da bitstring. L'insieme viene poi passato al blocco di post-elaborazione classica, dove viene utilizzata la tecnica di recupero della configurazione auto-consistente. Nel framework SQD, il ruolo del dispositivo quantistico è quello di produrre una distribuzione di probabilità.
Passaggio 1: Mappare il problema su un circuito quantistico
In questo tutorial, approssimeremo l'energia dello stato fondamentale di una molecola . Prima, specificheremo la molecola e le sue proprietà. Poi, creeremo un ansatz local unitary cluster Jastrow (LUCJ) (circuito quantistico) per generare campioni da un computer quantistico per la stima dell'energia dello stato fondamentale.
Prima, specificheremo la molecola e le sue proprietà.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import warnings
warnings.filterwarnings("ignore")
import pyscf
import pyscf.cc
import pyscf.mcscf
# Specify molecule properties
open_shell = False
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="6-31g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
num_orbitals = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
num_elec_a = (n_electrons + mol.spin) // 2
num_elec_b = (n_electrons - mol.spin) // 2
cas = pyscf.mcscf.CASCI(scf, num_orbitals, (num_elec_a, num_elec_b))
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), num_orbitals)
# Compute exact energy
exact_energy = cas.run().e_tot
converged SCF energy = -108.835236570775
CASCI E = -109.046671778080 E(CI) = -32.8155692383188 S^2 = 0.0000000
Successivamente, creeremo l'ansatz. L'ansatz LUCJ è un circuito quantistico parametrizzato, e lo inizializzeremo con le ampiezze t2 e t1 ottenute da un calcolo CCSD.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -109.0398256929734 E_corr = -0.2045891221988311
Utilizzeremo il pacchetto ffsim per creare e inizializzare l'ansatz con le ampiezze t2 e t1 calcolate sopra. Poiché la nostra molecola ha uno stato Hartree-Fock a shell chiusa, useremo la variante spin-bilanciata dell'ansatz UCJ, UCJOpSpinBalanced.
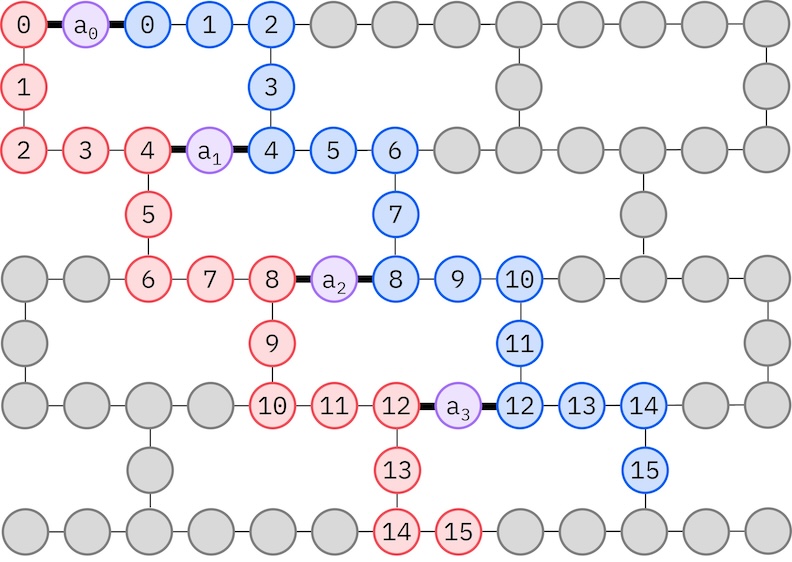
Poiché il nostro hardware IBM target ha una topologia heavy-hex, adotteremo il pattern zig-zag per le interazioni tra qubit. In questo pattern, gli orbitali (rappresentati da qubit) con lo stesso spin sono connessi con una topologia a linea (cerchi rossi e blu) dove ogni linea assume una forma zig-zag a causa della connettività heavy-hex dell'hardware target. Ancora, a causa della topologia heavy-hex, gli orbitali con spin diversi hanno connessioni ogni 4° orbitale (0, 4, 8, ecc.) (cerchi viola).
import ffsim
from qiskit import QuantumCircuit, QuantumRegister
n_reps = 1
alpha_alpha_indices = [(p, p + 1) for p in range(num_orbitals - 1)]
alpha_beta_indices = [(p, p) for p in range(0, num_orbitals, 4)]
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(alpha_alpha_indices, alpha_beta_indices),
)
nelec = (num_elec_a, num_elec_b)
# create an empty quantum circuit
qubits = QuantumRegister(2 * num_orbitals, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(num_orbitals, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Passaggio 2: Ottimizzare il problema
Successivamente, ottimizzeremo il nostro circuito per un hardware target. Dobbiamo scegliere il dispositivo hardware da usare prima di ottimizzare il nostro circuito. Useremo un backend fake a 127 qubit di qiskit_ibm_runtime per emulare un dispositivo reale. Per eseguire su una vera QPU, basta sostituire il backend fake con un backend reale. Consulta la documentazione di Qiskit IBM Runtime per maggiori informazioni.
from qiskit_ibm_runtime.fake_provider import FakeSherbrooke
backend = FakeSherbrooke()
Successivamente, raccomandiamo i seguenti passaggi per ottimizzare l'ansatz e renderlo compatibile con l'hardware.
- Selezionare i qubit fisici (
initial_layout) dall'hardware target che aderisce al pattern zig-zag descritto sopra. Disporre i qubit in questo pattern porta a un circuito efficiente compatibile con l'hardware con meno gate. - Generare un pass manager a stadi usando la funzione generate_preset_pass_manager di Qiskit con la tua scelta di
backendeinitial_layout. - Impostare lo stadio
pre_initdel tuo pass manager a stadi suffsim.qiskit.PRE_INIT.ffsim.qiskit.PRE_INITinclude i pass del Transpiler Qiskit che decompongono le gate in rotazioni orbitali e poi uniscono le rotazioni orbitali, risultando in meno gate nel circuito finale. - Eseguire il pass manager sul tuo circuito.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
spin_a_layout = [0, 14, 18, 19, 20, 33, 39, 40, 41, 53, 60, 61, 62, 72, 81, 82]
spin_b_layout = [2, 3, 4, 15, 22, 23, 24, 34, 43, 44, 45, 54, 64, 65, 66, 73]
initial_layout = spin_a_layout + spin_b_layout
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend, initial_layout=initial_layout
)
# without PRE_INIT passes
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/o pre-init passes): {isa_circuit.count_ops()}")
# with PRE_INIT passes
# We will use the circuit generated by this pass manager for hardware execution
pass_manager.pre_init = ffsim.qiskit.PRE_INIT
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/ pre-init passes): {isa_circuit.count_ops()}")
Gate counts (w/o pre-init passes): OrderedDict({'rz': 4420, 'sx': 3432, 'ecr': 1366, 'x': 239, 'measure': 32, 'barrier': 1})
Gate counts (w/ pre-init passes): OrderedDict({'rz': 2460, 'sx': 2156, 'ecr': 730, 'x': 71, 'measure': 32, 'barrier': 1})
Passaggio 3: Eseguire gli esperimenti
Dopo aver ottimizzato il circuito per l'esecuzione su hardware, siamo pronti a eseguirlo sull'hardware target e raccogliere campioni per la stima dell'energia dello stato fondamentale. Poiché abbiamo un solo circuito, useremo la modalità di esecuzione Job di Qiskit Runtime ed eseguiremo il nostro circuito.
Nota: Abbiamo commentato il codice per eseguire il circuito su una QPU e lo abbiamo lasciato come riferimento per l'utente. Invece di eseguire su hardware reale in questa guida, genereremo semplicemente campioni casuali estratti dalla distribuzione uniforme.
import numpy as np
from qiskit_addon_sqd.counts import generate_bit_array_uniform
# from qiskit_ibm_runtime import SamplerV2 as Sampler
# sampler = Sampler(mode=backend)
# job = sampler.run([isa_circuit], shots=10_000)
# primitive_result = job.result()
# pub_result = primitive_result[0]
# bit_array = pub_result.data.meas
rng = np.random.default_rng(24)
bit_array = generate_bit_array_uniform(10_000, num_orbitals * 2, rand_seed=rng)
Passaggio 4: Post-elaborare i risultati
Ora, eseguiamo l'algoritmo SQD usando la funzione diagonalize_fermionic_hamiltonian. Consulta la documentazione API per le spiegazioni degli argomenti di questa funzione.
Il solver incluso nell'addon SQD usa l'implementazione di PySCF del CI selezionato, nello specifico pyscf.fci.selected_ci.kernel_fixed_space. L'esempio seguente mostra anche come passare argomenti keyword a quella funzione tramite il solver incluso. Qui passiamo l'argomento max_cycle.
from functools import partial
from qiskit_addon_sqd.fermion import SCIResult, diagonalize_fermionic_hamiltonian, solve_sci_batch
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 1
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}")
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=num_orbitals,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -105.45358671756313
Subspace dimension: 5476
Iteration 2
Subsample 0
Energy: -107.95172900082163
Subspace dimension: 249001
Iteration 3
Subsample 0
Energy: -108.97460330369815
Subspace dimension: 339889
Iteration 4
Subsample 0
Energy: -109.02739376648793
Subspace dimension: 440896
Iteration 5
Subsample 0
Energy: -109.030972328451
Subspace dimension: 597529
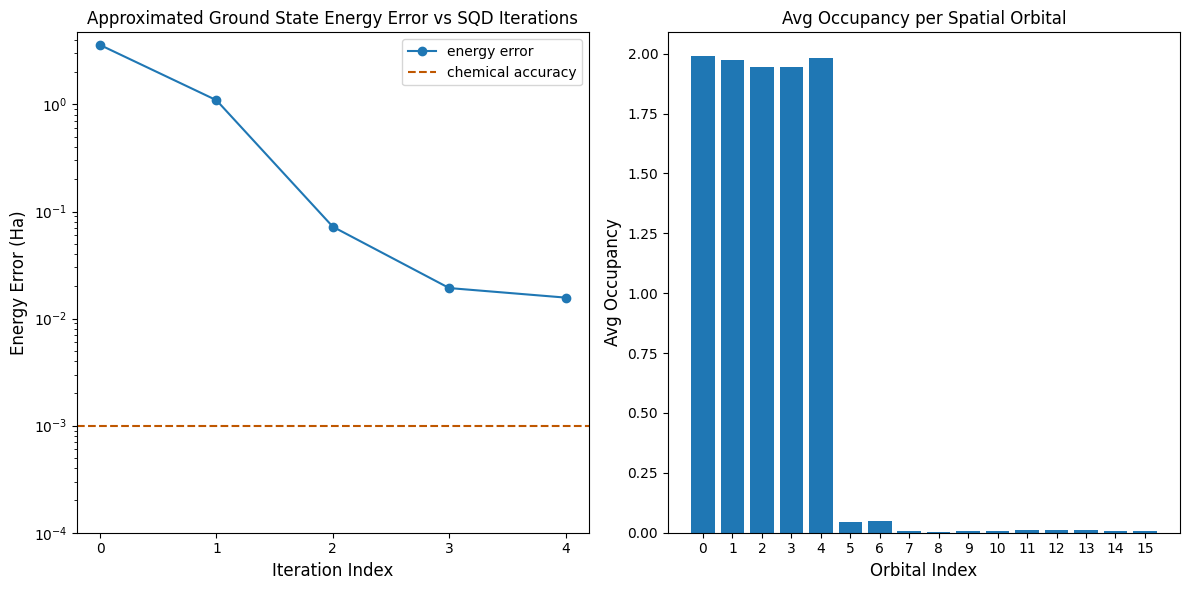
Ora, tracciamo i risultati.
Il primo grafico mostra che dopo alcune iterazioni stimiamo l'energia dello stato fondamentale entro ~16 mH (la precisione chimica è tipicamente accettata come 1 kcal/mol 1.6 mH). Ricorda, i campioni quantistici in questa demo erano puro rumore. Il segnale qui proviene dalla conoscenza a priori della struttura elettronica e dell'Hamiltoniano molecolare.
Il secondo grafico mostra l'occupazione media di ciascun orbitale spaziale dopo l'iterazione finale. Possiamo vedere che sia gli elettroni con spin-up che quelli con spin-down occupano i primi cinque orbitali con alta probabilità nelle nostre soluzioni.
import matplotlib.pyplot as plt
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - exact_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(y=chem_accuracy, color="#BF5700", linestyle="--", label="chemical accuracy")
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Exact energy: {exact_energy:.5f} Ha")
print(f"SQD energy: {min_e[-1]:.5f} Ha")
print(f"Absolute error: {e_diff[-1]:.5f} Ha")
plt.tight_layout()
plt.show()
Exact energy: -109.04667 Ha
SQD energy: -109.03097 Ha
Absolute error: 0.01570 Ha